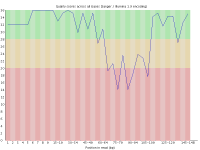
With the introduction of the NextSeq system Illumina changed the way their image data was acquired so that instead of capturing 4 images per cycle they needed only 2. This speeds up image acquisition significantly but also introduces a problem where high quality calls for G bases can be made where there is actually no signal on the flowcell.
May 4, 2016
Simon Andrews
NextSeq, All Applications, Cutadapt, FastQC
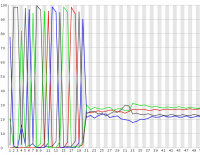
In some experimental designs a large proportion of the sequences in a library can have identical sequence at their 5′ end. These types of library can cause problems for the data collection and base calling on illumina sequencers, leading to the generation of poor quality data.
March 15, 2016
Simon Andrews
Illumina, All Applications, FastQC
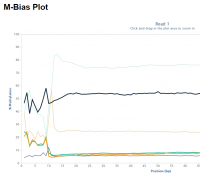
Random priming in PBAT libraries introduces drastic biases in the base composition and methylation levels especially at the 5′ end of all reads. As a result, affected bases should be removed from the libraries before the alignment step.
March 11, 2016
Felix Krueger
Illumina, Methylation, PBAT, BamQC, Bismark, FastQC, Trim Galore!
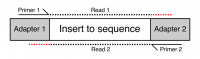
Many sequencing platforms require the addition of specific adapter sequences to the end of the fragments to be sequenced. For an individual fragment, if the length of the sequencing read is longer than the fragment to be sequenced then the read will continue into the adapter sequence on the end. Unless it is removed this adapter sequence will cause problems for downstream mapping, assembly or other analysis.
February 7, 2016
Simon Andrews
Cutadapt, FastQC, Skewer, Trim Galore!

The assumption when analysing sequence datasets is that every sequence comes from a different biological fragment in the original sample. Many library preparation techniques though include one or more PCR steps which introduce the possibility that the same original fragment can be observed multiple times, biasing the results produced. In some cases this type of duplication can be extreme and have a serious effect on the ability to analyse the data correctly.
February 6, 2016
Simon Andrews
FastQC, MultiQC, Preseq, SeqMonk

The construction of sequencing libraries on many platforms requires the addition of specific adapter sequences to the ends of the fragments to be sequenced. Although there are steps in place to ensure that only valid adapter+insert combinations make it onto the sequencer it is possible to get adapter dimers with no valid insert making it through the sequencing process.
February 1, 2016
Simon Andrews
Illumina, FastQC
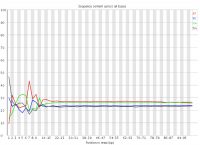
In a randomly primed library there is no reason to expect that a specific sequencing cycle should contain more of one specific base than any other cycle. It is commonly observed though that this type of library does contain a cycle-specific sequence bias, most frequently in the initial bases of the run.
January 31, 2016
Simon Andrews
mRNA-Seq, FastQC

Rather than a general loss of quality for a whole sequencing lane or run sometimes partial failures occur. These can affect specific regions and cycles and can have knock on effects for the data generated
January 31, 2016
Simon Andrews
Illumina, FastQC
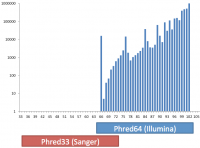
Base call qualities in fastq and BAM format files are recorded using a text based encoding. Mistakes in the way this encoding is applied can lead to incorrect interpretation of base call accuracies in downstream analyses.
January 20, 2016
Simon Andrews
FastQC, QC Software
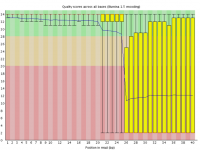
Sometimes a sequencing run can experience a sudden and lasting loss of base call quality across all sequences.
January 20, 2016
Simon Andrews
Illumina, FastQC, QC Software
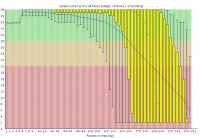
Illumina based sequencing shows a loss of base call quality as the number of sequencing cycles performed increases.
January 20, 2016
Simon Andrews
Illumina, FastQC, QC Software