Introduction
In 2012, Miura and colleagues described a new method of preparing libraries for bisulfite sequencing termed Post-Bisulfite Adapter Tagging (PBAT). In this technique DNA is sheared by the bisulfite reaction itself, and the fragments that are later sequenced are regenerated by two rounds of random priming with random tetrameric (4N) oligos and strand extension. The undisputed advantage of this method is that a lot less starting material is needed which makes it amenable for rare cell types or even single-cell analyses. Several optimised PBAT-type protocols have since been published or are even available as commercial kits, e.g. the EpiGnome (epicentre) or Pico Methyl-Seq (Zymo) kits.
We and others have noticed early on however that most of these methods introduce a hefty bias in sequence composition at the 5′ end of reads, which corresponds in length exactly to the length of the oligo used for random priming (4N in the original paper, but methods often also use 6N, 9N or 12N). All examples shown in this article used a 9N oligo for the initial pulldown reaction.
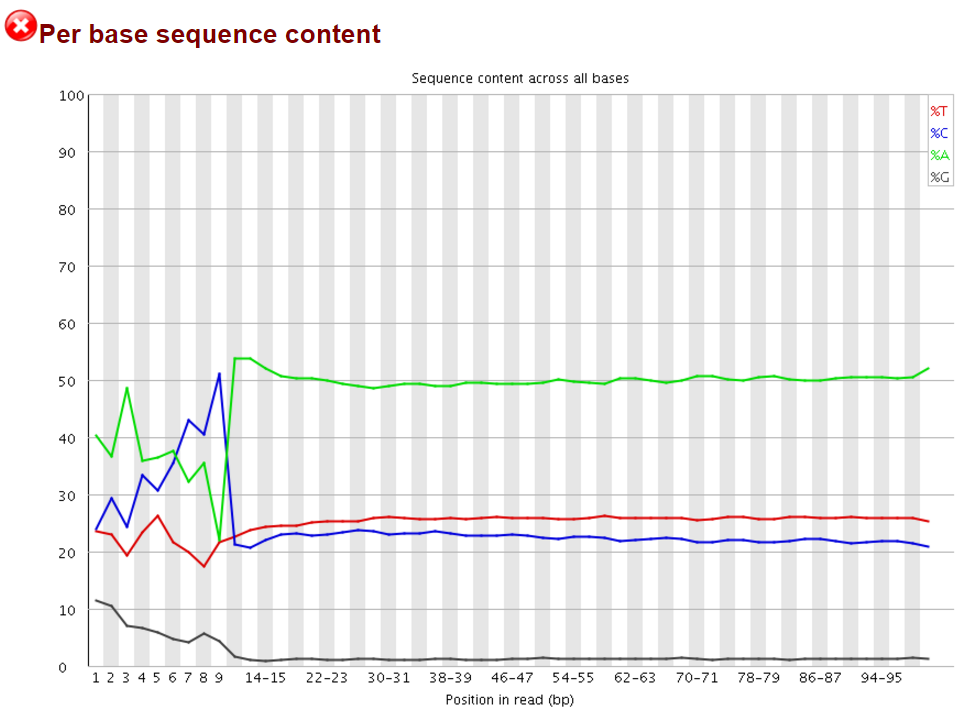
The Symptoms
The biased sequence composition is also carried through to the aligned sequences (not shown here), and sure enough the methylation levels at the first couple of positions is also severely distorted, especially in non-CG context, as can be seen from the Bismark M-bias plot (please also see the M-bias plot in this post):
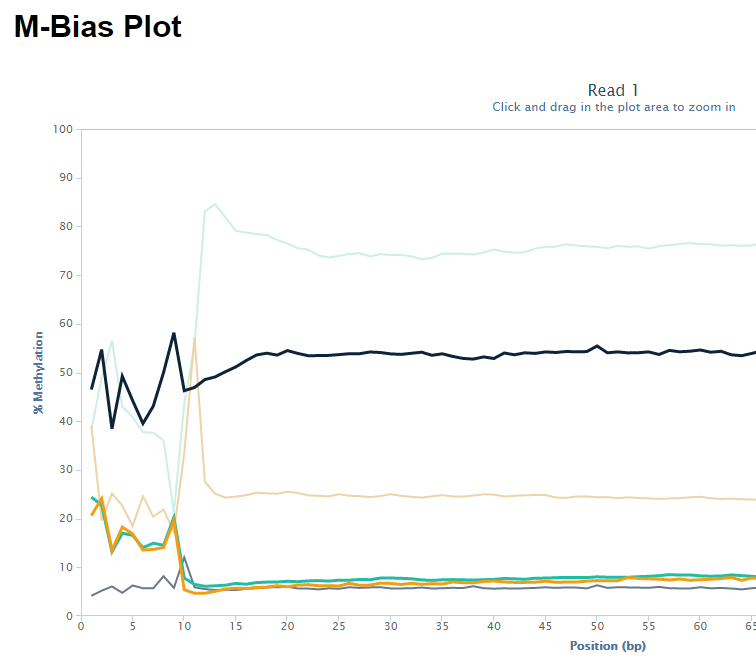
We never really got to root cause of the problem but were convinced that this has to be a technical artefact rather than a biological effect. We suggested to simply remove the first bases either by 5′ trimming the raw sequences, or by ignoring the first N bases during the methylation extraction.
Diagnosis
5′ trimming the 4N, 6N or 9N portion of single-end (SE) reads works like a charm for both mapping and M-bias plots, the data looks reasonably clean afterwards and one can achieve mapping rates of 60-70% for 100bp reads and mapping to the mouse genome (which is fairly good).
It gets a bit more tricky for paired-end (PE) reads however, because simply removing the biased positions (marked as B below) for both reads of paired-end alignments may result in ‘dovetailing’ reads, where one or both reads seem to extend past the start of the mate read. Bowtie2 alignments in Bismark are carried out invariably using the option --no-discordant so that really odd combinations of read pairs (such as oriented away from each other, only one read aligns, reads align to different chromosomes etc.) are not reported as valid paired-end alignments. Dovetailing alignments are also considered discordant and thus do not get reported.
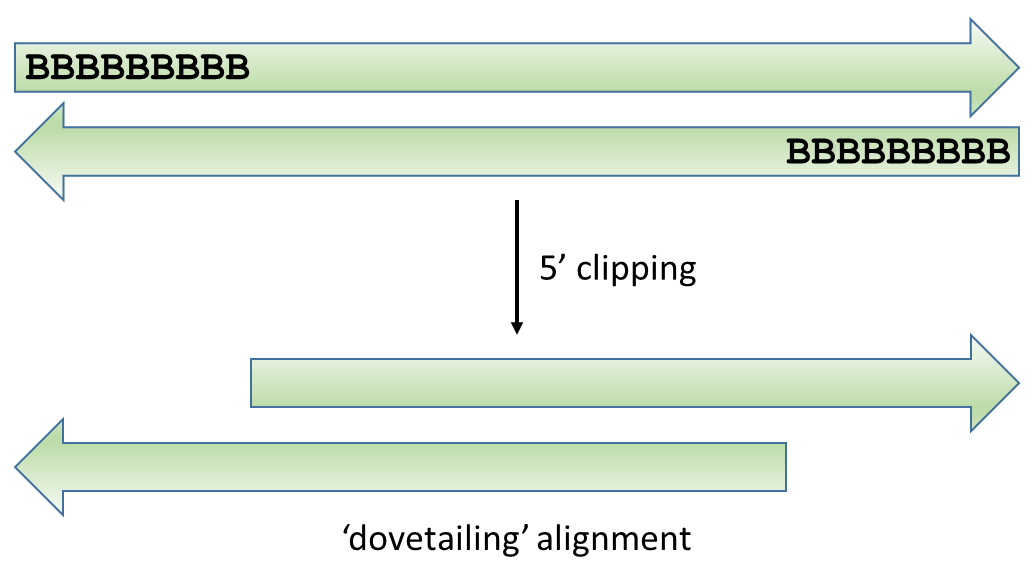
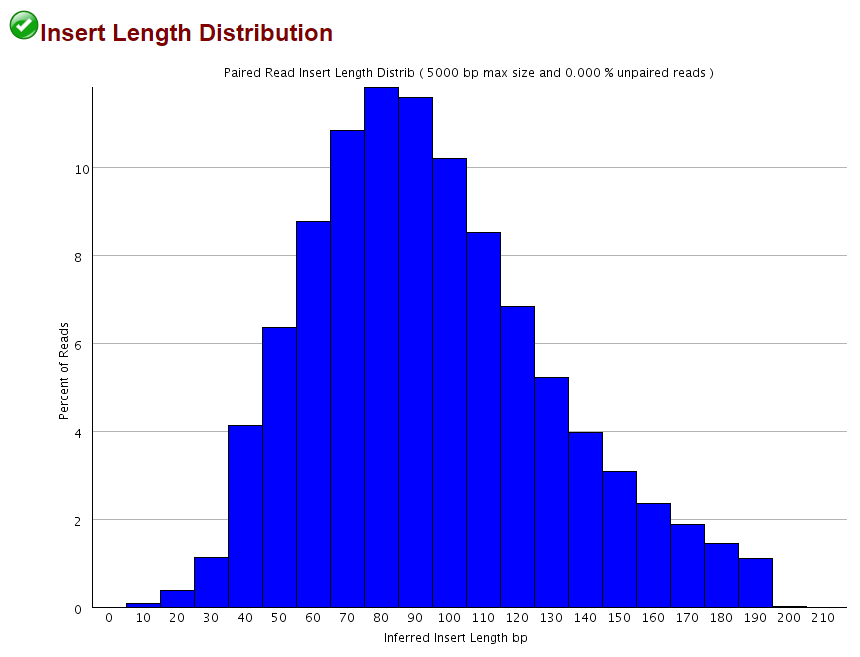
This obviously only applies when the sequenced fragment is shorter than the sequencing read length, but as shown for the 2×102 bp (9N PBAT) example above there are quite a few alignments shorter than 100bp:
Since we knew that dovetailing reads were a problem we tended to run PE alignments without 5′ clipping the biased positions (just doing the usual adapter and poor quality removal using Trim Galore) and instead ran the methylation extractions with the options --ignore 9 --ignore_r2 9 to disregard the methylation calls from the first couple of positions. This effectively removes the horrible bias at the start but people were complaining about poor mapping efficiencies…
To find out whether we should align the data including the first positions (the argument being that they were genuine captured genomic sequences) or remove them before trimming we ran alignments with 5′ unclipped data and then looked at the rate of SNPs within the first couple of positions of the aligned data:
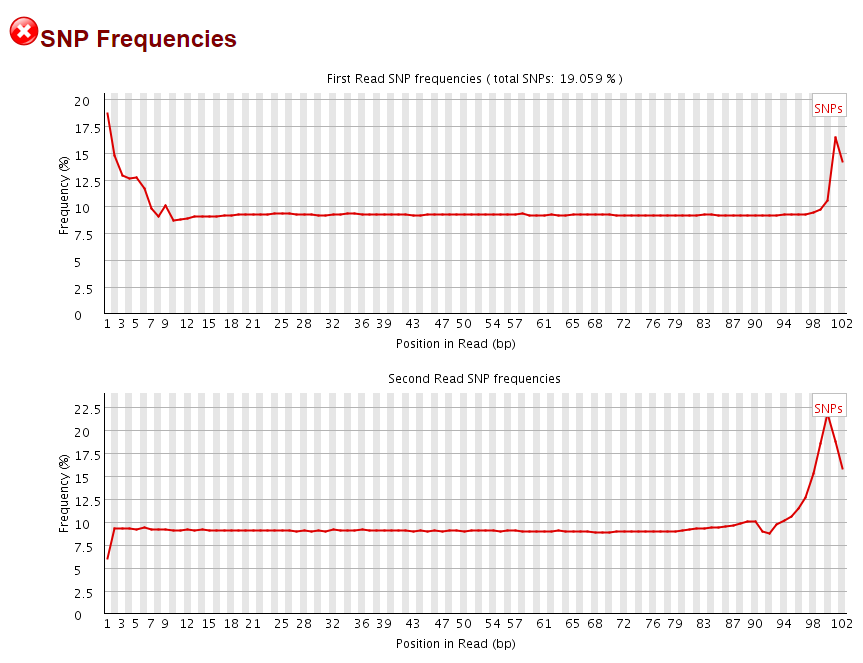
It was quite intriguing to see that the level of SNPs found in the first 9bp of Read 1 and last 9bp of Read 2 (corresponding to the start of R2) were drastically elevated. It has to be mentioned that the overall frequency of SNPs appears fairly high due to the presence of C->T conversions that are mere methylation changes rather than true SNP calls.
In addition the reads also sported a sharply increased frequency of insertions or deletions at the start of Read 1 and Read 2, again corresponding almost perfectly to the 9N random primed portion of the reads:
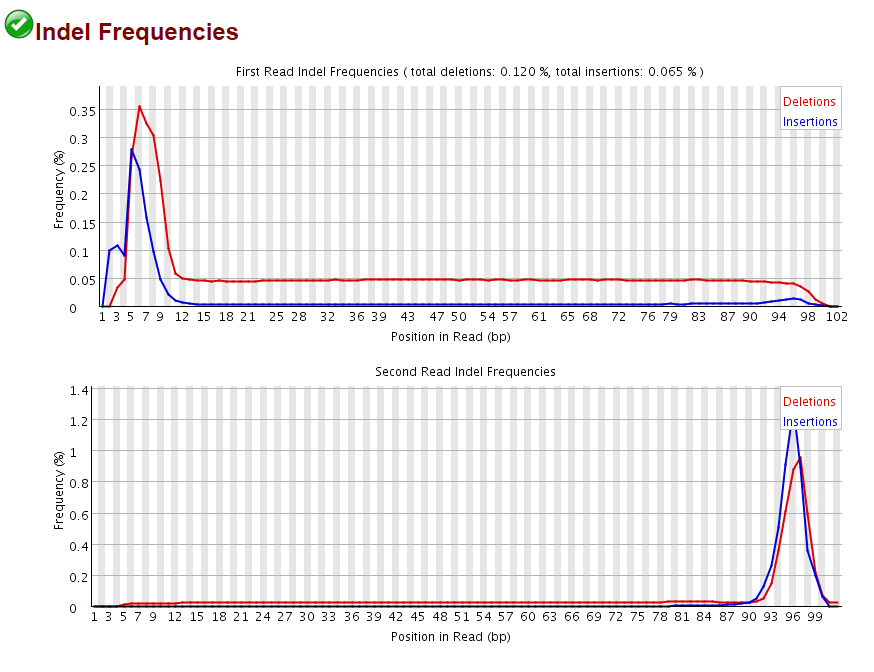
This demonstrated convincingly that there are weird things going on during the “random priming” step of the PBAT protocol, resulting in a drastically increased rate of SNPs and InDels. Since both SNPs and InDels make it more difficult to map a read we reasoned that this could be at least in part responsible for the poor mapping efficiencies observed for 5′ unclipped PE PBAT libaries. When the alignments were run again relaxing the mapping parameters (allowing three times as many mismatches and/or indels than in the default setting) the mapping efficiency did indeed increase dramatically from 39 to 58% (not shown). This demonstrated nicely that this library was largely comprised of valid paired-end alignments, but since we expect that a much increased rate of mismatches and indels at the start will also result in incorrect methylation calls we do not recommend relaxing the mapping parameters to remedy the low mapping efficiency of PE PBAT alignments.
Mitigation
To get around this issue we implemented a new option --dovetail into Bismark so that dovetailing alignments are now considered concordant. This allows us to run Trim Galore on the data specifying the length of the random pulldown oligos (here 9 bp) as parameter for 5′ clipping, e.g. with a command like this:
trim_galore --clip_R1 9 --clip_R2 9 --paired sample_R1.fastq.gz sample_R2.fastq.gz
This of course auto-detects and removes read through adapter contamination and poor basecall qualities as well. Running test alignments with Bismark/Bowtie2 and default parameters with either i) unclippped regular alignments, ii) 9 bp 5′ clipped regular alignments or iii) 9 bp 5′ clipped alignments with --dovetail gave the following results:
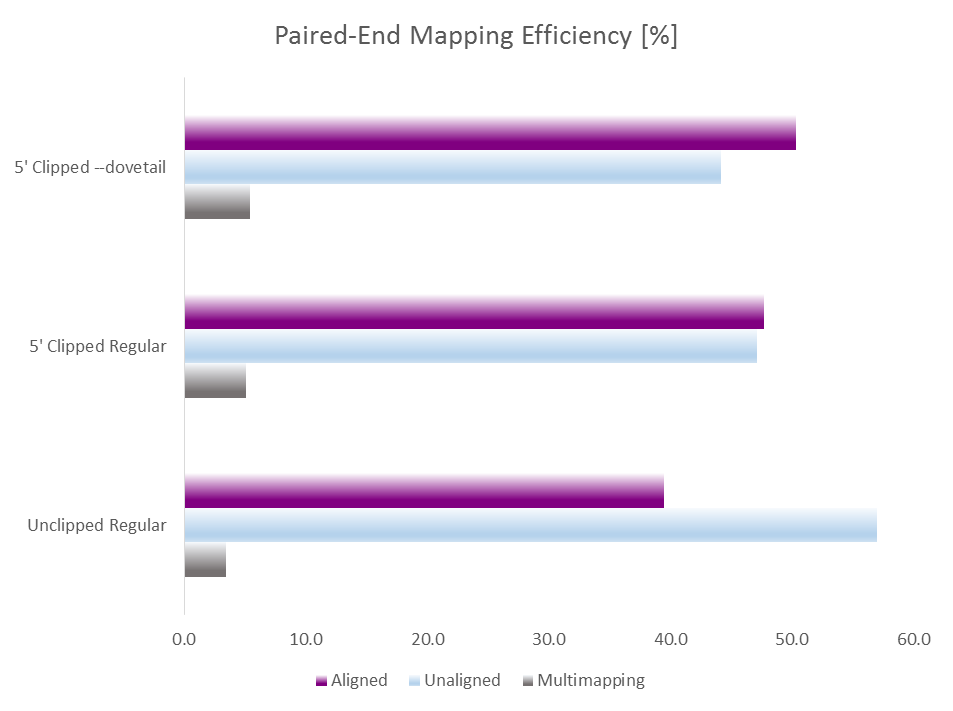
Merely clipping 9bp of the 5′ end already increased the mapping efficiency from 39 to 47% which is in line with removing lots of inhibitory indel and SNP errors within the 9N random priming part. Allowing dovetailing alignments with --dovetail increased the mapping efficiency further to > 50% while at the same time lowering the rate of (probably contaminating) non-CG methylation levels (not shown).
As a result of this the option --dovetail will now be turned by default whenever --pbat is selected in Bismark (version 0.15.1 and higher, available from the Bismark GitHub page).
Prevention
As long as there are no major changes in the PBAT protocol the problems described in this article are unlikely to go away. We do recommend 5′ trimming of the random priming portion of reads in addition to removing standard read-through adapters and poor qualities before aligning the data. Specifying --pbat in Bismark will automatically set --dovetail as well so the default mode should do just right thing from now on.
A word about hybrid reads in PBAT libraries
While the measures described above should be pretty effective in combating errors introduced by the PBAT protocol it should be noted that we have seen a tendency of hybrid reads in PBAT libraries. Hybrid reads in this context are reads that would be considered discordant, i.e. where R1 and R2 align to completely different places in the genome. These reads will not be reported by Bismark as valid alignments, but we have seen paired-end libraries with in excess of 30% of all fragments aligning as to different places in the genome similar to a bisulfite Hi-C experiment (should there be one…). It is debatable whether a read pair starting at say chromosome 3 and continuing into chromosome 17 contains credible methylation information, but clearly a fairly large portion of hybrid reads seems to contribute to the generally low-ish mapping efficiency observed for some PE PBAT libraries.
As a solution to this problem we would probably recommend to run 5′ clipping and trimming first, and run PE alignments with --unmapped specified in Bismark. The resulting unmapped Read 1 and Read 2 files may then be aligned in single-end mode afterwards to salvage as much data from alignable hybrid reads in the experiment as possible (--pbat for R1 and default settings for R2).
Lessons Learnt
I think we have now finally got to the bottom what causes the weird biases in PBAT-type libraries. It is important that the community understands that the problems associated with the biased positions and takes appropriate measures (a typical lane of HiSeq may potentially result in several (hundred) million incorrect methylation calls otherwise…).
Side Note – Bias on the 3′ end
One could argue that the bias described above exists on both sides of fragments because the 3′ end is going to be filled in by a copy mechanism from the other, biased, strand. While this is probably true it seems that elimination of the bias on the 5′ end is much more relevant because of the following reasons:
- the 5′ bias affects potentially all reads
- the 3′ bias is only sequenced if the fragment length is shorter than the read length. Depending on the library preparation this may only be a small subset of the total library
- we don’t see a distinct increase of SNPs, indels or M-bias changes towards the end of reads, even though this could be masked by different the read ending at different positions. (The peak in SNPs for Read 1 in the example above probably stems from a rather low amount of sequences with that length, and might in fact be caused by adapter trimming)
- the 3′ end is subject to adapter/quality trimming, and thus for short enough fragments the bias may have been removed already by the quality trimming step
- the 3′ bias of Read 2 would not be counted anyway because as soon as reads start overlapping the methylation calls of R2 are ignored during the methylation extraction
So in summary, at the current time we believe that only ever half (R1) of a subset (short) of a subset (not quality trimmed) of total reads might affected by a bias on the 3′ end. In other words, it might be a source of error one can live with.
References
Original paper describing the PBAT method. Miura et al., 2012.
Datasets
No dataset specifically, but any PBAT library we have seen so far exhibited the same problems. Just find one on GEO.
Software
QC plots were generated with FastQC or BamQC.
Adapter/quality trimming and 5′ clipping was performed with Trim Galore.
Bisulfite reads were aligned and plots generated using Bismark (on GitHub).