
For speed and efficiency many RNA-Seq mapping protocols map either entirely or initially to a transcriptome sequence. When the sample is contaminated with genomic material you can get significant numbers of reads reported as uniquely mapping when they actually map to many locations within the genome.
February 2, 2016
Simon Andrews
mRNA-Seq, SeqMonk

In theory RNA-Seq samples only contain reads derived from RNA. Most protocols include a DNase treatment to remove any remaining DNA from the sample. It is fairly common however to see samples with significant amounts of DNA contamination. Ignoring this can bias the counts generated from the data and throw off normalisation.
February 2, 2016
Simon Andrews
RNA-Seq, SeqMonk
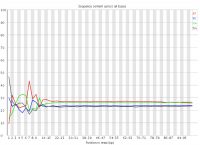
In a randomly primed library there is no reason to expect that a specific sequencing cycle should contain more of one specific base than any other cycle. It is commonly observed though that this type of library does contain a cycle-specific sequence bias, most frequently in the initial bases of the run.
January 31, 2016
Simon Andrews
mRNA-Seq, FastQC