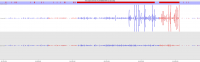
Soft-clipping of sequencing reads allows the masking of portions of the reads that do not align to the genome from end to end, which may be desirable for certain types of analysis (e.g. detection of structural variants). For standard alignment processes soft-clipping may however incorrectly trim reads and lead to the mis-assignments of reads primarily to repetitive regions. This phenomenon appears to vary in severity for different sequencing applications with Bisulfite sequencing being worst off.
May 16, 2016
Felix Krueger
All Applications, Bismark, Bowtie2, bwa-meth, HISAT2, SeqMonk, Trim Galore!
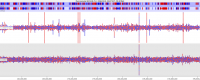
With the increasing capacity of a single flowcell lane it can be tempting to mix samples of different types within the same lane to make the most of your sequencing, but cross contamination between libraries in a flowcell can lead to the generation of artefacts which can mess up your analysis.
April 15, 2016
Simon Andrews
Illumina, All Applications, SeqMonk

Paired-end libraries generated by Post Bisulfite Adapter Tagging (PBAT) often suffer from poorer mapping efficiencies when compared to standard whole genome shotgun Bisulfite-Seq libraries. In addition to the usual suspects that have a detrimental impact on mapping efficiency we found that a substantial proportion of paired-end PBAT libraries appears to consist of chimeric reads that map to different places in the genome, not unlike Hi-C type experiments. Chimeric reads also affect single-cell libraries (scBS-seq) as they are constructed using a PBAT approach.
March 18, 2016
Felix Krueger
Illumina, Methylation, PBAT, Bismark, Cutadapt, SeqMonk, Trim Galore!

One of the standard fields in the SAM/BAM file format is the mapping quality (MAPQ) value. This value can be very useful to help filter mapped reads before doing downstream analysis – unfortunately the implementation of this value is in no way consistent between different aligners so it takes a fair bit of research to know how to use it appropriately. Mis-applying the filter could cause reads to be inappropriately excluded from an analysis.
March 17, 2016
Simon Andrews
All Technologies, All Applications, BamQC, SeqMonk
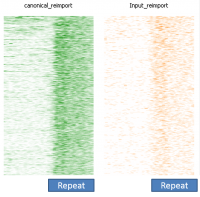
In a dataset where you have some degree of technical duplication, and have not filtered your data to only keep uniquely mapping reads then if you perform deduplication it will look as if repeat sequences are enriched.
February 11, 2016
Simon Andrews
ChIP-Seq, Data Processing, SeqMonk

The assumption when analysing sequence datasets is that every sequence comes from a different biological fragment in the original sample. Many library preparation techniques though include one or more PCR steps which introduce the possibility that the same original fragment can be observed multiple times, biasing the results produced. In some cases this type of duplication can be extreme and have a serious effect on the ability to analyse the data correctly.
February 6, 2016
Simon Andrews
FastQC, MultiQC, Preseq, SeqMonk

For speed and efficiency many RNA-Seq mapping protocols map either entirely or initially to a transcriptome sequence. When the sample is contaminated with genomic material you can get significant numbers of reads reported as uniquely mapping when they actually map to many locations within the genome.
February 2, 2016
Simon Andrews
mRNA-Seq, SeqMonk

In theory RNA-Seq samples only contain reads derived from RNA. Most protocols include a DNase treatment to remove any remaining DNA from the sample. It is fairly common however to see samples with significant amounts of DNA contamination. Ignoring this can bias the counts generated from the data and throw off normalisation.
February 2, 2016
Simon Andrews
RNA-Seq, SeqMonk