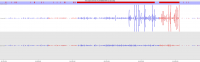
Soft-clipping of sequencing reads allows the masking of portions of the reads that do not align to the genome from end to end, which may be desirable for certain types of analysis (e.g. detection of structural variants). For standard alignment processes soft-clipping may however incorrectly trim reads and lead to the mis-assignments of reads primarily to repetitive regions. This phenomenon appears to vary in severity for different sequencing applications with Bisulfite sequencing being worst off.
May 16, 2016
Felix Krueger
All Applications, Bismark, Bowtie2, bwa-meth, HISAT2, SeqMonk, Trim Galore!

Paired-end libraries generated by Post Bisulfite Adapter Tagging (PBAT) often suffer from poorer mapping efficiencies when compared to standard whole genome shotgun Bisulfite-Seq libraries. In addition to the usual suspects that have a detrimental impact on mapping efficiency we found that a substantial proportion of paired-end PBAT libraries appears to consist of chimeric reads that map to different places in the genome, not unlike Hi-C type experiments. Chimeric reads also affect single-cell libraries (scBS-seq) as they are constructed using a PBAT approach.
March 18, 2016
Felix Krueger
Illumina, Methylation, PBAT, Bismark, Cutadapt, SeqMonk, Trim Galore!
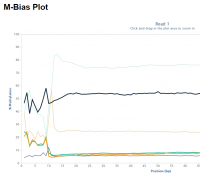
Random priming in PBAT libraries introduces drastic biases in the base composition and methylation levels especially at the 5′ end of all reads. As a result, affected bases should be removed from the libraries before the alignment step.
March 11, 2016
Felix Krueger
Illumina, Methylation, PBAT, BamQC, Bismark, FastQC, Trim Galore!
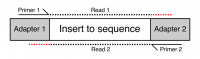
Many sequencing platforms require the addition of specific adapter sequences to the end of the fragments to be sequenced. For an individual fragment, if the length of the sequencing read is longer than the fragment to be sequenced then the read will continue into the adapter sequence on the end. Unless it is removed this adapter sequence will cause problems for downstream mapping, assembly or other analysis.
February 7, 2016
Simon Andrews
Cutadapt, FastQC, Skewer, Trim Galore!