Introduction
Many types of sequencing library incorporate some kind of random fragmentation in order to generate the fragments which go on to be sequenced. Looking at the per sequencing cycle sequence content and checking that there isn’t a positional bias is a standard part of many QC platforms. In some experimental designs though you can have a situation where a very high proportion of the library (possibly all of it) will at least start with exactly the same sequence. This type of library structure can lead to problems data collection and base calling on illumina sequencing platforms.
The Symptoms
A per-base sequence composition plot from such a biased composition library is shown below. As you can see all reads in this library start with the same initial sequence, but then after 20 bases the sequences diverge.
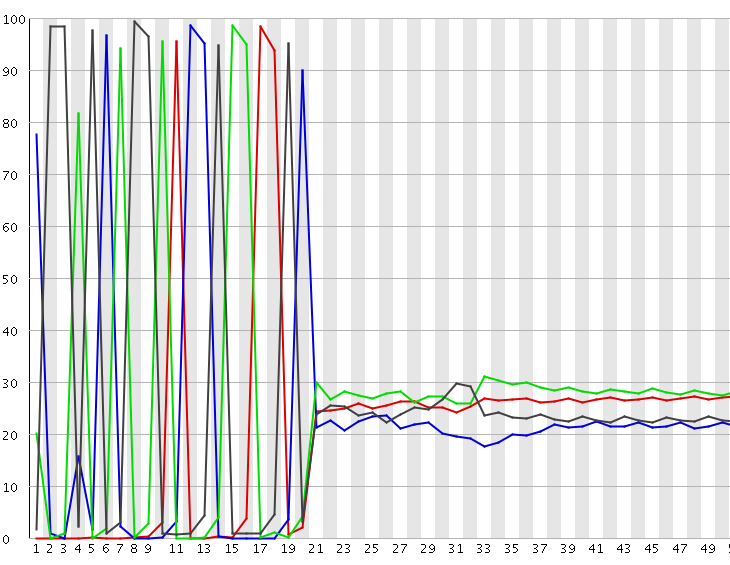
In this type of library the symptoms observed will be any or all of:
- Poor overall sequence qualities
- Low cluster numbers, with a high number of clusters being rejected during the run
- A high incidence of ‘N’ calls in the output
- Higher than expected positional bias within the flowcell
Depending on the severity of the problem you may even get an almost complete failure of the run, although more normally you will get reduced yield with poorer quality calls.
Diagnosis
In order to understand the reason for these failures it is necessary to explain some of the steps in the illumina data analysis pipeline which relate to this problem. There are a few steps in the data collection which can be affected by these types of libraries:
1) Focussing
Illumina sequencing is an imaging based data collection. You have a glass flowcell onto which clusters of DNA molecules are attached, and after the addition of tagged nulceotides there is an imaging step where a computer controlled microscope images the surfaces of the flowcells. Right at the start of the run one of the critical steps in the data collection is to set the focussing for this flowcell. This focussing has to be done before the first data collection, so at the point of focussing only the first base in each cluster will be visible. The illumina system uses a number of different flurophore molecules and lasers to measure the different bases, but for focussing it just picks one of these channels to use, the idea being it doesn’t need to see every cluster, it just needs enough information to see the correct focal plane. If however you have little or no data in the channel being used to set the focussing then there is effectively nothing for the instrument to focus on, and it’s possible that the initial focussing will be so far out that it will never recover, generating out of focus clusters for the rest of the run with very bad knock on effects for data quality. The instrument does have the ability to adjust focus during the run, but this refocussing is limited to small changes, so if the initial focus is too far out it may not recover completely.
2) Cluster detection
On most illumina systems DNA clusters are randomly positioned over the surface of the flowcell (this is changing in more recent systems with the introduction of semi-ordered arrays of clusters) – so one of the other initial steps in processing is to identify where each individual cluster is on the flowcell so that it can be tracked through the run to produce an individual output sequence. When putting clusters on the flowcell there are two competing factors which need to be taken into account – ideally you’d like each cluster to be completely isolated from each other cluster so that as soon as you see a signal in the imaging you can assume that it comes from a single DNA sequence. However you also want to put as many clusters on the flowcell as possible to maximise the amount of data you get from a run. In practice therefore flowcells are loaded to a point where a significant proportion of the clusters will in contact with one or more other clusters such that a simple identification of the position of signals in the first sequencing cycle is not enough to identify the position of every individual cluster.

The way this problem is resolved is that cluster calling is not done solely on the data from the first sequencing cycle – instead data is collected from the first few cycles and each identified spot is analysed. If in any cycle a spot shows a pattern where, for example, the left side of the spot is mostly G signal, but the right side of the spot is mostly C signal then the system can assume that this is actually two spots which are touching and can treat the two halves as separate spots for future analysis.
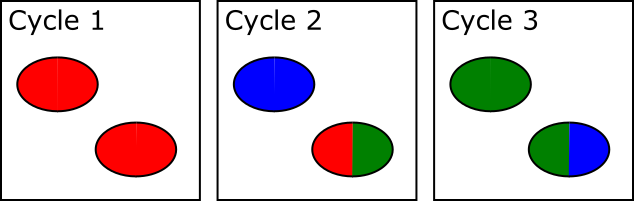
In the example below, both spots shown are actually two adjacent and touching clusters. The one at the bottom right can be distinguished as two clusters since the sequence within the two sub-areas changes within the set of cycles used for cluster detection, whilst the one at the top left has the same sequence through early cycles and will be analysed incorrectly as if they were a single cluster.
The problem with this approach is that there is a limit to how far into the run the system can detect these merged spots. After a spot is identified as being merged the system must go back to the raw image data for the two halves of that spot from the start of the run to call signal for the two regions separately. This means that until spot detection is complete all of the raw image data for the run must be kept, and this data is BIG! Practically therefore the spot detection can only run for a few cycles. For biased libraries the effect of this is that unless the library sequence for two overlapping spots diverges within the set of cycles used for cluster detection it will be considered as a single cluster for the remainder of the run. When the sequence eventually diverges it will appear as a mixed signal and the cluster will likely be discarded due to this.
3) Channel calibration
The final aspect to this type of failure is the calibration of the specific parameters used to base call each run. For optimal base calling the software needs to work out the relative strengths observed in the different measured channels in each run. This accounts for variations in the laser or detector efficiencies or in the properties of the fluors used in the libraries. Setting these calibtration values must, by design, make some assumptions about the nature of the nature of the data it expects to see, so when a library is hugely biased (possibly to the point where some channels are completely blank) it is perhaps not surprising if the paramters for the run are not set optimally, and base call quality is therefore reduced.
Mitigation
To some extent this problem has been greatly mitigated by improvements in the base calling software by illumina. Improvements in the focussing and calibration now mean that libraries rarely completely fail (which used to be a common occurrence on the GA sequencers), but loss of sequences, inclusion of N’s in the calls, and lower than normal phred scores are still common. Informatics improvements can only takcle some of these issues, ultimately some of them require changes in the libraries or sequencing reactions to fix.
Some common fixes to this problem are:
- You can spike diverse sequence into the library. Most commonly this will be the PhiX control library which Illumina themselves provide. Even a low (~5%) amount of this will give enough signal to fix the focussing and will improve the calibration. Higher amounts will begin to alleviate the cluster detection issue as you’re more likely to see overlaps between a fixed sequence and PhiX, but this obviously comes at the cost of wasting sequencing capacity on generating phix sequence.
- You can restructure your library to not have a fixed sequence at the front. You can improve some aspects of the sequencing by adding a random barcode to the front of the insert, but fixed bases later in the library will still be a problem. If the random sequence can be made to be variable length then this will provide diversity for the rest of the library, and as long as you can identify the start of your insert this can provide a more complete solution.
- If your library has a completely fixed sequence at the start which later turns into diverse sequence then the easiest and most effective fix is to change the primer used for the sequencing to one which primes immediately upstream of the diverse region. This has a couple of advantages, it stops you wasting sequencing capacity sequencing the common sequence, and it means the library starts at a diverse position so the calibration and cluster detection will be good.

Lessons Learnt
Sometimes a good understanding of the methodology of your sequencing platform can help to understand sequencing failures and point to possible solutions.
References
Krueger F, Andrews SR, Osborne CS Large Scale Loss of Data in Low-Diversity Illumina Sequencing Libraries Can Be Recovered by Deferred Cluster Calling PLoS One. 6(1): e16607.