Introduction
A sequencing library consists of a set of DNA fragments for which sequence is to be obtained. Many sequencing platforms cannot sequence the DNA directly, but require the addition of platform specific adapter sequences to the ends to provide anchorage and priming points for the sequencing chemistry reactions. The sequencing reaction is designed so that the primer will anneal immediately upstream of the insert to be sequenced, so the first sequence to be generated is the desired insert sequence, and not the adapter.
In some cases the length of the inserts in the library might be quite short – short enough that in some cases they are shorter than the read length of the sequencing run. Where the read length exceeds the length of the insert then the read will run off the end of the desired sequence and into the adapter on the other end. This will lead to the addition of non-native sequence on the end of some reads in the library. These will appear at various positions within the length of the reads, but will always consist of the same sequence. In the diagram below the red regions of the reads would be read-through adapter.
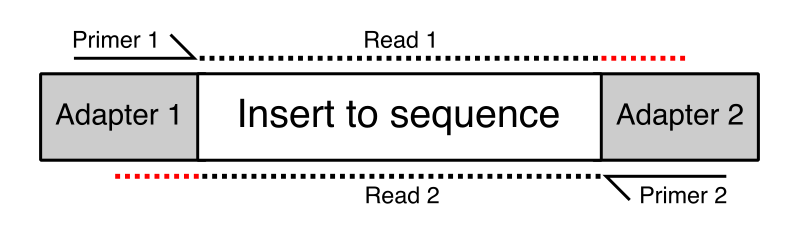
The Symptoms
The addition of artificial adapter sequence on the end of reads in a library will normally cause those reads to fall out of any downstream analysis fairly quickly. The bases in the adapter sequence will generally have high phred scores as there is no reason for the sequence to be of poor quality. They will introduce large numbers of mismatches into any genomic alignments causing affected reads not to map to reference genomes. Assembly with these reads will likewise fail since proper extension will also fail at the point where adapter sequence is reached.
Diagnosis
Since the sequence of the adapters used for a given library is known ahead of time, and that sequence is normally not present then it is possible to explicitly screen for that sequence within the library to identify which reads contain adapter and at what distance into the read. Since the number of different adapters used by the major platforms is relatively small it is even possible to screen for all of the normal adapter sequences as a matter of course. This will both check for the presence of adapters in the library and also confirm that it is the expected adapter which has been found.
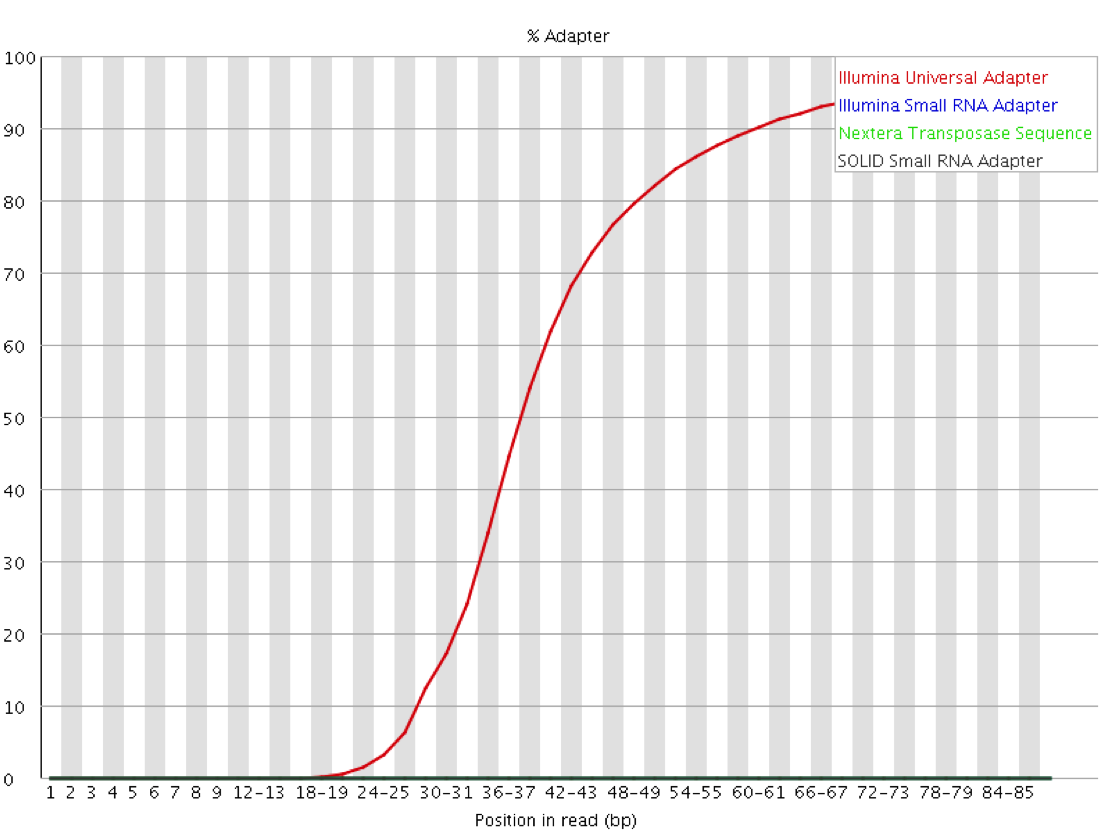
Mitigation
Adapter trimming is the standard approach to removing adapter sequence from sequencing libraries. In this process a search is performed for the adapter sequence within each read, and the read is truncated to the point where the adapter is found to start. This kind of trimming is easy when there is a substantial amount of adapter present since there is only a low likelihood that this could be native sequence. It becomes more difficult where the added length of adapter is relatively short (only a few bases) since small numbers of bases consistent with being adapter could easily be present on the end of a native sequence. There are a few options in these cases:
- Aggressive adapter trimming – you can choose to remove even the shortest amounts of adapter from the end of all reads. This should provide fairly complete removal of adapter sequence, but will also trim back native sequence where the end of the read coincidentally looks like short amounts of adapter. This kind of aggressive trimming may be preferred where small amounts of adapter could have a deleterious effect on downstream analysis, such as for SNP detection or for detection of methylation in bisulphite sequencing. This kind of trimming will also introduce a sequence bias into the end of libraries which can trigger warnings in other QC. For example if the first base of an adapter is a T then no full length sequence will be allowed to end with a T since this could represent a 1bp adapter read-through so you should see a complete depletion of T in the last base of the full read length.
- Limited adapter trimming – you can set a threshold for the amount of adapter which must be present before being trimmed. Requiring the presence of several bases of adapter which continue to the end of the read will reduce the amount of over-trimming you perform, but will mean that some short adapter regions will remain in the library. If the nature of the library is such that the mismatches from these short adapter stretches could be significant then you might want to use a more aggressive trimming strategy.
- If you have performed paired end sequencing then you can use the information from the other end of the pair to decide on the exact position of the adapter. If you sequence long enough to read right through the insert in read 1, then you would read past the point where read 2 would start from (but on the opposite strand). In these cases you can actually align reads 1 and 2 and this will allow you to determine the exact extent of the true insert, which will run from the start of read 1 to the start of read 2.
Prevention
The only way to prevent adapter read through would be to either make the inserts longer, or to reduce the length of the sequencing reads performed. On many platforms there is a practical limit to how long insert sequences can be, and in many cases short inserts reflect underlying biology, so the need for adapter removal may well be around for the foreseeable future.
Software
The presence of read through adapters can be screened for with FastQC. There are many trimming programs available which all operate in roughly similar ways. We use (and wrote) trim galore which is designed to automatically detect the correct adapter to trim by looking into the data. It itself is a wrapper around the cutadapt program which does the actual trimming.
The detection and removal of unknown adapters in paired end data can be performed with skewer.
One thought on "Read-through adapters can appear at the ends of sequencing reads"
Long Wang
I have noticed some adapters could appear in the middle of reads from Hiseq platforms… No one knows why …
Comments are closed