
Probably the single biggest problem with the mapping of reads to a reference sequence is dealing with reads which come from parts of the genome which aren’t in the assembly. These reads can cause significant amounts of noise in anlayses performed on genomic data.
March 21, 2016
Simon Andrews
All Technologies, All Applications

One of the standard fields in the SAM/BAM file format is the mapping quality (MAPQ) value. This value can be very useful to help filter mapped reads before doing downstream analysis – unfortunately the implementation of this value is in no way consistent between different aligners so it takes a fair bit of research to know how to use it appropriately. Mis-applying the filter could cause reads to be inappropriately excluded from an analysis.
March 17, 2016
Simon Andrews
All Technologies, All Applications, BamQC, SeqMonk
We are increasingly re-using data deposited in public sequence archives such as SRA, ENA or DDBJ and we rely on being able to successfully extract data from these sources. In some cases we have found that errors in the validation of the data can mean that data is corrupted when it is downloaded from these repositories.
March 3, 2016
Simon Andrews
All Technologies, All Applications
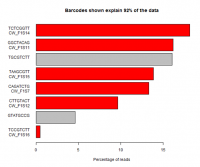
When running multiple samples in the same sequencing reaction the different libraries are usually created with unique sequence tags (barcodes) to allow the sequence from the lane to be separated. Problems during this splitting are common and can have serious effects on downstream analysis.
February 12, 2016
Simon Andrews
All Technologies, All Applications, Data Processing