Introduction
In any library which has been generated as the result of random fragmentation or random priming there should be no reason to expect that the base composition in a given cycle should be different to any other. The overall composition of the library might different between runs (GC content of the source species, enrichment of certain bases, chemical modifications of bases etc), but these should affect all cycles equally. It is a general tenet of QC that the clear observation of structure in datasets which are supposed to be unstructured should be a cause for concern – yet we often see fairly strong positional sequence biases at the start of random primed libraries, so should this be a cause for concern?
The Symptoms
The problem described here is mostly clearly seen in a per-base sequence content plot. A typical example of an RNA-Seq library affected by this issue is shown below:
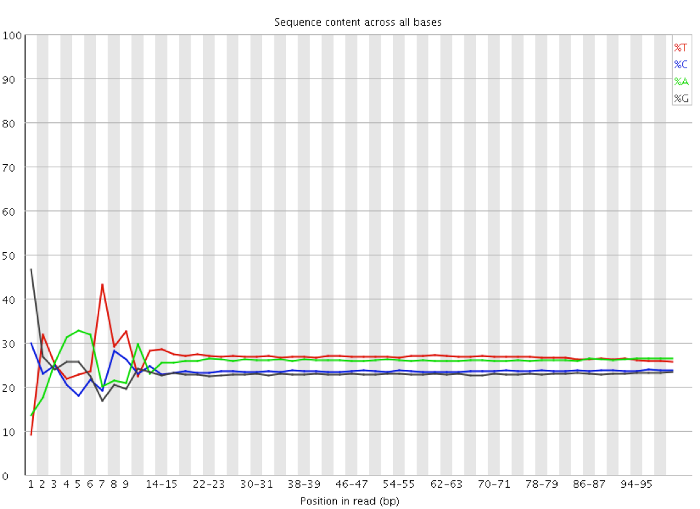
You can clearly see the biased sequence in the first ~12 bases of the run. This bias then dissipates over the rest of the run which shows the expected parallel tracks in the base content for each base. This happens in pretty much all RNA-Seq libraries to a greater or lesser extent.
The cause of this bias it turns out is the random priming step in library production. The priming should be driven by a selection of random hexamers which in theory should all be present with equal frequency in the priming mix and should all prime with equal efficiency. In the real world it turns out that this isn’t the case and that certain hexamers are favoured during the priming step, resulting in the based composition over the region of the library primed by the random primers.
The question then arises as to whether this bias has any implications for downstream analyses. There are a couple of potential concerns:
- It’s possible that there is increased mis-priming as part of the bias – introducing an increased number of mis-called bases at the start of the sequence
- It’s possible that the selection bias introduced will have a significant effect of the ability of the library to fairly measure the content of the original RNA population due to certain sequences being unduly favoured
In practice it seems that neither of these concerns is really a problem. The bias at the start of the sequences appears to be the result of biased selection of fragments from the library, so high levels of predicted SNPs are not an issue. The biased selection though doesn’t appear to be strong enough to cause major headaches in downstream quantitation of data. A strong bias would result in a very uneven coverage of different parts of a transcript based on its sequence content, and most RNA-Seq libraries do not show these types of localised biases (excepting biases from mappability and other factors beyond this effect). Also the biases are very similar between libraries, so any artefacts which were introduced should cancel out when doing any kind of differential analysis.
Diagnosis
This problem is most easily detected with the FastQC per-base sequence content plot.
Mitigation
People often suggest fixing this issue by 5′ trimming of the reads to remove the biased portion – this however is not a fix. Since the biased composition is created by the selection of sequencing fragments and not by base call errors the only effect of trimming would be to change from having a library which starts over biased positions, to having a library which starts slightly downstream of biased positions.
Prevention
Ultimately this only fix for this issue will be in the introduction of new library preparation kits with a less bias prone priming step.
Lessons Learnt
Whilst the warnings generated by this problem reflect a real issue it’s not something which can be fixed, and doesn’t seem to have any serious consequences for downstream analysis. Ironically if you are producing RNA-Seq libraries it would make for better QC if you were to focus on libraries which didn’t have this artefact in them, as they would be the ones which were truly suspicious.
References
Biases in Illumina transcriptome sequencing caused by random hexamer priming.
Hansen KD, Brenner SE, Dudoit S.
Nucleic Acids Res. 2010 Jul;38(12):e131. doi: 10.1093/nar/gkq224. Epub 2010 Apr 14.
9 thoughts on "Positional sequence bias in random primed libraries"
James Hadfield
I’m not sure if the paper you referenced checked to see if all the data they used was Illumina RNA-Seq? It might be the case that the bias is slightly different with other kit providers using different random hexamer mixes.
Yannick Wurm
My xp with RNAseq is limited. But the following graphs suggests to me that more may be going on. If the positional sequence bias is due only to non-randomness of primers, then you wouldn’t expect to see any effect on mapping to the reference.
However, in our recent NextSeq run we do find that that STAR mapper has an increased likelihood of clipping the 5′ end of the read. This suggests that there may additionally be an effect of biased basecalling (which could obviously be higher with 2-color chemistry).
Images:
http://www.antgenomes.org/~yannickwurm/tmp/fastqcACGT.png
http://www.antgenomes.org/~yannickwurm/tmp/star_qualimap_clipping.png
Simon Andrews
For some positional biases (we’ve seen this mostly in BS-Seq libraries), the sequence bias is also accompanied by an increased rate of mutation. Our assumption in these cases is that a slightly mis-primed primer is being used and is artificially creating a variant which wasn’t in the original sequence. There may be variation in this depending on the nature of the random priming set and the conditions used for annealing.
Yannick Wurm
(although there is also a chance that clipping could occur during mapping if we’re on a splice junction and the aligner has insufficient information to confidently map to the previous exon)
Simon Andrews
Soft clipping has its own set of issues – there’s a whole other article about that!
Andrés
Hi,
I wonder if this bias could affect transcriptome de novo assemblers. I couldn’t find any paper that had tested it.
Have a nice day
Salva
I have found the 5′ bias in my RNA-seq data from a wild reptile.
Now, I am interested in both expression profiles (leading to a physiological study) and full reconstruction of heat-shock genes which are unknown for my study species (leading to a phylogenetic study).
Although I can see that the 5′ bias should not affect expression profiles, I would appreciate your view on whether trimming should be a must if I want to reconstruct a set of specific genes.
Many thanks.
Salva
Gopal
Hi, is this normal to see this effect in genome sequencing data as well? As it happens I see exactly the same effect in Illumina DNA sequencing data that I currently have and wondering if that needs to be clipped.
Thomas van Gurp
We have documented the mispriming of random hexamers previously: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0085583