Introduction
Illumina sequencing chemistry works by tagging a growing DNA strand with a labelled base to indicate the last nucleotide which was added. 4 different dyes are used for the 4 different bases and imaging the flowcell after each base addition allows the machine to read the most recently added base for each cluster.
In the standard version of this system used on HiSeq and MiSeq instruments each cycle of chemistry is followed by the acquisition of 4 images of the flowcell, using filters for each of the 4 emission wavelenths of the dyes used. The time taken for this imaging is significant in the overall run time, so it would obviously be beneficial to reduce this.
With the introduction of the NextSeq system Illumina introduced a new imaging system which reduced the number of images per cycle from 4 to 2. They did this by using filters which could allow the simultaneous measurement of 2 of the 4 dyes used. By combining the data from these two images they could work out the appropriate base to call.
4 colour detection
| Base | G filter | A filter | T filter | C filter |
|---|---|---|---|---|
| G | Yes | No | No | No |
| A | No | Yes | No | No |
| T | No | No | Yes | No |
| C | No | No | No | Yes |
2 colour detection
| Base | A+C filter | T+C filter |
|---|---|---|
| G | No | No |
| A | Yes | No |
| T | No | Yes |
| C | Yes | Yes |
The problem here though is that there is a fifth base option, which is that there is no signal there to detect. This could happen for a number of reasons:
- There is no priming site for this read, so no extension is happening. If this happens in the first read then it just won’t detect a cluster, but in subsequent reads the cluster will be assumed to still be present.
- Enough of the cluster has degraded or stalled that the signal remaining is too faint to detect
- Something is physically blocking the imaging of the flowcell (air bubbles, dirt on the surface etc etc)
In the 4 colour system this extra option is easy to identify since you get no signal from anything, but in the 2 colour system we have a problem, since no signal in either channel is what you’d expect from a G base, so you can’t distinguish no signal from G.
What we find therefore on Illumina systems running this chemistry is that we see an over-calling of G bases in these cases, and this can cause problems in downstream processing of data.
The Symptoms
There are a couple of different ways in which this problem can manifest itself. Possibly the most obvious is that you can get a huge over-representation of poly-G sequences in reads other than the first read (which is used for cluster detection). From reports we’ve seen this seems to be most prevalent in the first barcode read, but could presumably affect any read where the initial priming of the read failed for any reason.
The example below is the top of a barcode splitting report for a sample which had a lot of barcodes. You can see that the most frequently observed barcode for this sample had polyG for barcode1, and that this wasn’t expected. You can also see that the second barcode was read correctly so the cluster itself was OK, but for some reason just didn’t prime for the barcode 1 read.
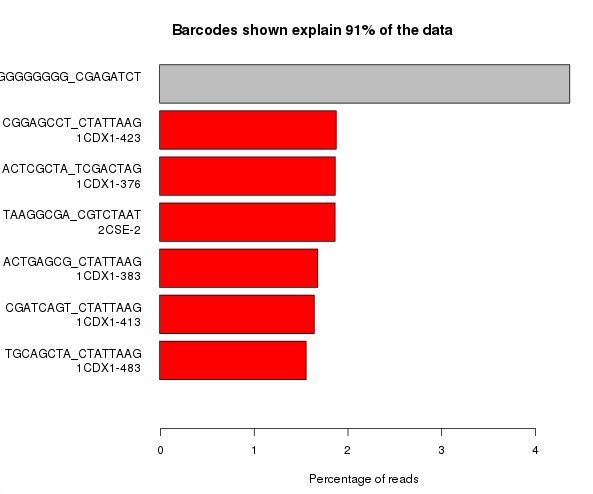
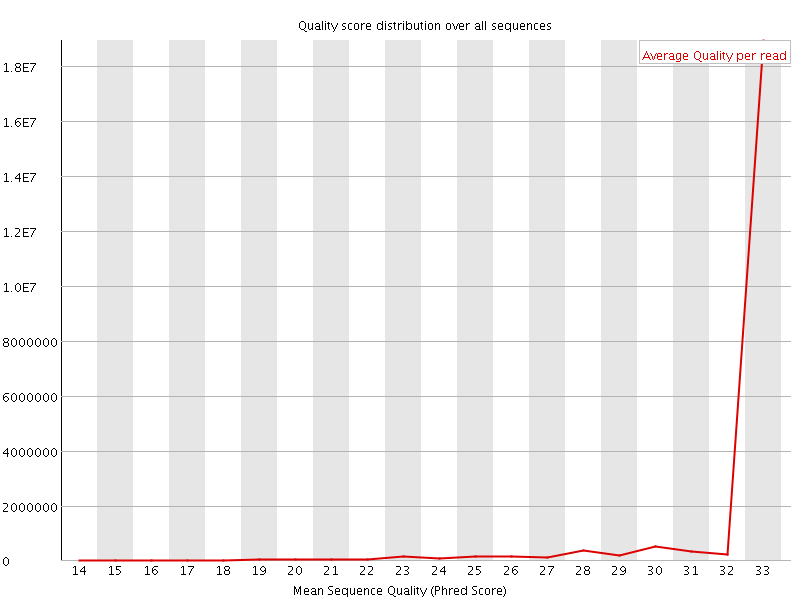
If we extract out the polyG sequences from the first barcode read and look at their quality distribution we see that the qualities are universally high, so there’s no way to distinguish these mis-calls by using the normal quality control mechanism.
This then leads to the second type of problem seen in these datasets. This occurs when the signal from a cluster completely degrades. In a 4 colour system this would result in a somewhat random base call, but a very low quality score to reflect the low level of signal. In a 2 colour system the quality will initially fall as the signal degrades, but eventually the signal will effectively disappear and the sequencer will start calling high quality G bases.
The sequence below shows this effect:
@1:11101:2930:2211 1:N:0 ATTTATTATTAATTAAATATTAATAATAAATAGATCGGAAGAGCACACGTCTGAACTCCAGTCACTAGCTTAGCGCGTATGCCGTCGTCGGCGTGCAAAAAAAAAGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG +AAAAAEEEEEEEEEEEE6EEEEEAEEEEEEEEEA/EE<EEEAEE/EAEEAEEEE6</EEEEEA/<//<///A/A//////</E<//////E///A/</A/<<A////A/E<EEEEEEEAEEE/EEEAEAEAEAE6/AEAEE<AAEAEE
It’s easier to see if you visualise the quality scores for this sequence
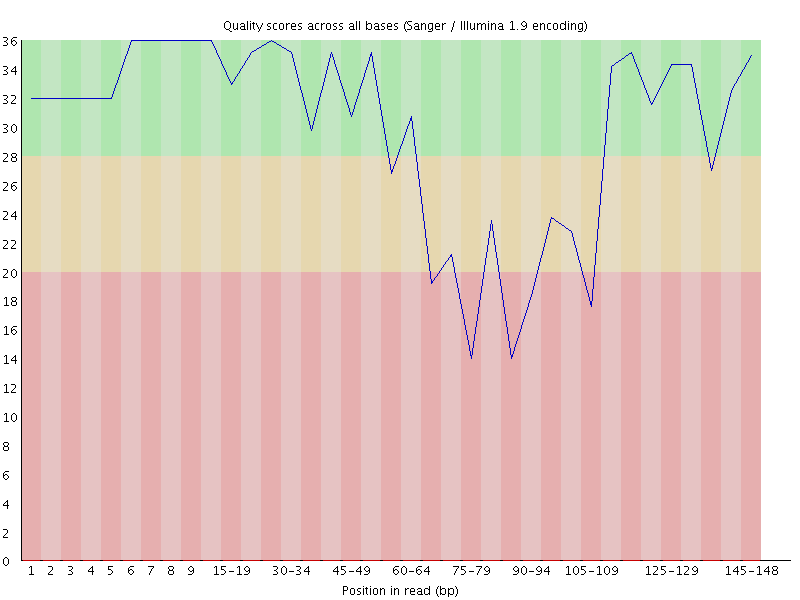
You can see that the quality degrades as expected initially, but then the signal disappears, either because of an extension of the general degradation or because some external factor blocked the imaging, and the sequencer starts to call G bases, the quality rises again.
The big issue this cases is that most trimming systems assume that quality loss is progressive from the 5′ end of a read to the 3′, so when deciding where to quality trim a read they start at the 3′ end and work back towards the 5′ until good quality sequence is detected. In the types of read shown above the read will not be trimmed because the most 3′ sequence is high quality, so incorrect base calls will remain which will adversely affect the downstream analysis of this data.
Mitigation and Prevention
There are a few potential strategies for fixing this problem and some of the symptoms will be easier than others. For unprimed reads which are completely polyG then these will probably not affect downstream analyses other than triggering QC alerts, so will have less of an impact once people are aware of their source. For the progressive quality loss the need for a solution is more pressing. Illumina already have a system in place which artificially downweights the quality scores for reads whose quality dips below a certain level. It seems likely that they should be able to adapt their calling software to recognise reads where quality dips to be replaced with high quality poly-G and then artificially downweight the quality scores for the remainder of the read. This same case can probably also be tacked by the authors of trimming programs where they might, for example, provide and option to exempt G calls from the assessment of quality and to trim 3′ Gs from reads as a matter of course.
Lessons Learnt
You can’t always trust the quality scores coming from sequencers, and if you see odd sequences in your libraries it’s worth investigating them as they may unearth a deeper problem.
Acknowledgements
Many thanks to Hemant Kelkar, Marcela Davila Lopez, Stuart Levine and Frederick Tan for their help in understanding this issue.
4 thoughts on "Illumina 2 colour chemistry can overcall high confidence G bases"
Marcel Martin
I’ve recently added a –trim-nextseq option to cutadapt to deal with this issue. It’s marked as experimental, but works well on the test data I had. You currently need to install cutadapt from the Git repository to get the option; it will be part of the next release.
Joe Foley
Illumina’s TruSeq LT (single-index) barcode A023 is GAGTGG. This is only two edits away from no signal, which is what you’ll see for a cluster that lost its index read or the PhiX control, which has no barcode. Anecdotally, I had a problem once that looked like a lot of NextSeq reads from a library with barcode A023 getting misassigned as PhiX in a custom demultiplexing pipeline. Has anyone else noticed problems with A023 on the NextSeq? My result isn’t conclusive but there’s an obvious design problem, so I’ve just avoided using that combination of index and platform ever since.
Simon Andrews
We do custom splitting for our barcodes and after trying a few different regimes we now only assign a read if the barcode is a full and exact match to the expected sequence. We probably lose a few reads this way, but we save ourselves a bunch of headaches from mis-assigning reads to the wrong sample precisely in cases like this.
Ólavur Mortensen
Has this problem been addressed in NextSeq? We exclusively do run NextSeq 500 runs, how can I check if our data has this problem?
Comments are closed