The new Illumina patterned flow cell technology uses chemistry that is prone to “index hopping”, leading to reads being assigned to the wrong sample in multiplexed sequencing runs. This problem has already caused countless research groups to re-assess their experimental results or refrain from using the new sequencers. However, with careful experimental preparation it may be possible to ameliorate this situation so as to become negligible for most applications
June 7, 2017
Steven Wingett
HiSeq, Illumina, X Ten, All Applications
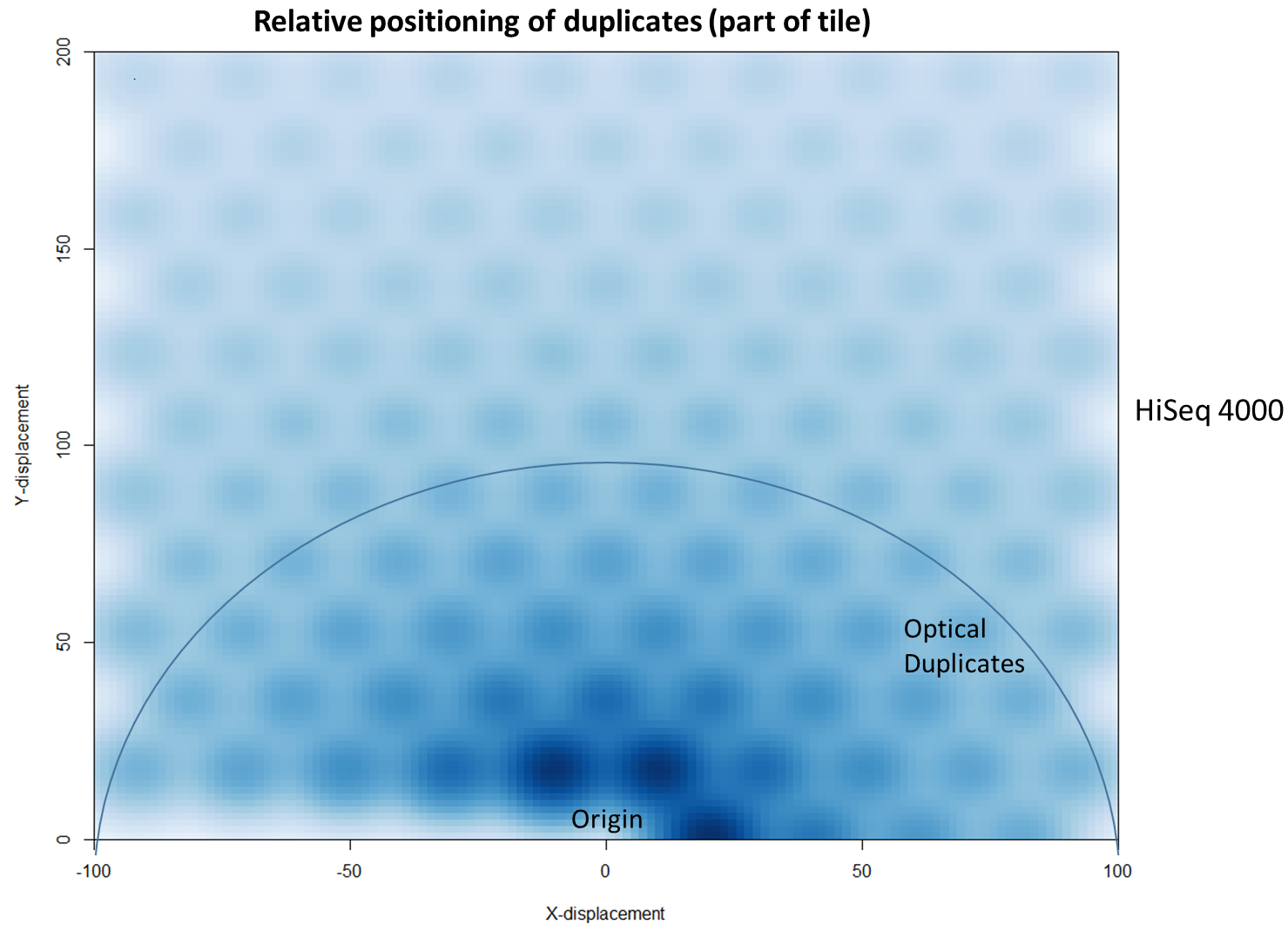
The latest Illumina sequencers – such as the HiSeq X, HiSeq 3000 and HiSeq 4000 – use patterned flow cells to enable the discrimination between much more densely packed DNA clusters. While such technology substantially increases the number of reads generated per sequence run, this innovation may lead to an increased number of duplicates, thereby negating the improved yield and making subsequent data analysis potentially more difficult. Further investigation shows that these putative sequencing duplicates are generally in close two-dimensional proximity on a flow cell, which may provide an opportunity to develop bioinformatics solutions to identify and discard such artefacts.
March 2, 2017
Steven Wingett
HiSeq, All Applications, Bowtie2, HiCUP, Picard
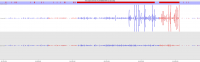
Soft-clipping of sequencing reads allows the masking of portions of the reads that do not align to the genome from end to end, which may be desirable for certain types of analysis (e.g. detection of structural variants). For standard alignment processes soft-clipping may however incorrectly trim reads and lead to the mis-assignments of reads primarily to repetitive regions. This phenomenon appears to vary in severity for different sequencing applications with Bisulfite sequencing being worst off.
May 16, 2016
Felix Krueger
All Applications, Bismark, Bowtie2, bwa-meth, HISAT2, SeqMonk, Trim Galore!
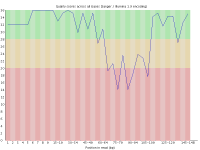
With the introduction of the NextSeq system Illumina changed the way their image data was acquired so that instead of capturing 4 images per cycle they needed only 2. This speeds up image acquisition significantly but also introduces a problem where high quality calls for G bases can be made where there is actually no signal on the flowcell.
May 4, 2016
Simon Andrews
NextSeq, All Applications, Cutadapt, FastQC
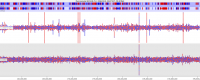
With the increasing capacity of a single flowcell lane it can be tempting to mix samples of different types within the same lane to make the most of your sequencing, but cross contamination between libraries in a flowcell can lead to the generation of artefacts which can mess up your analysis.
April 15, 2016
Simon Andrews
Illumina, All Applications, SeqMonk

Probably the single biggest problem with the mapping of reads to a reference sequence is dealing with reads which come from parts of the genome which aren’t in the assembly. These reads can cause significant amounts of noise in anlayses performed on genomic data.
March 21, 2016
Simon Andrews
All Technologies, All Applications

One of the standard fields in the SAM/BAM file format is the mapping quality (MAPQ) value. This value can be very useful to help filter mapped reads before doing downstream analysis – unfortunately the implementation of this value is in no way consistent between different aligners so it takes a fair bit of research to know how to use it appropriately. Mis-applying the filter could cause reads to be inappropriately excluded from an analysis.
March 17, 2016
Simon Andrews
All Technologies, All Applications, BamQC, SeqMonk
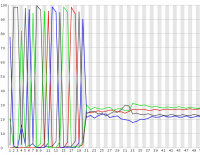
In some experimental designs a large proportion of the sequences in a library can have identical sequence at their 5′ end. These types of library can cause problems for the data collection and base calling on illumina sequencers, leading to the generation of poor quality data.
March 15, 2016
Simon Andrews
Illumina, All Applications, FastQC
We are increasingly re-using data deposited in public sequence archives such as SRA, ENA or DDBJ and we rely on being able to successfully extract data from these sources. In some cases we have found that errors in the validation of the data can mean that data is corrupted when it is downloaded from these repositories.
March 3, 2016
Simon Andrews
All Technologies, All Applications
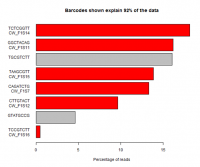
When running multiple samples in the same sequencing reaction the different libraries are usually created with unique sequence tags (barcodes) to allow the sequence from the lane to be separated. Problems during this splitting are common and can have serious effects on downstream analysis.
February 12, 2016
Simon Andrews
All Technologies, All Applications, Data Processing