Introduction
A single lane on an Illumina sequencer can generate in excess of 200 million reads – far in excess of what is needed for many common sequencing applications. It is therefore common to put multiple samples in the same lane, and to use barcode sequences to be able to determine the original source for each read so you can separate them later on.
The most common use of barcoding is to mix together multiple samples from the same experiment, where all of the samples will be of the same type and from the same species, but we have frequently seen cases where people want to mix very different libraries together. Whilst we have strongly recommended against this for a long time (and don’t even allow samples to be annotated that way in our LIMS), people persist in doing this so it seemed worthwhile illustrating why this is a bad idea.
The root problem here is that barcoding is not a foolproof technology. Even with stringent separation criteria (all barcodes must be read exactly correctly over their entire length), you will still get a small amount of cross contamination between the libraries within a single lane. Whilst this will be a randomly selected subset of reads and will represent only a fraction of a percent of the total data, this can still have a devastating effect on the analysis of some library types and is very difficult to fix retrospectively.
The Symptoms
This issue is well illustrated in by the following genome view of two ChIP-Seq lanes which are ChIPing for the same factor.
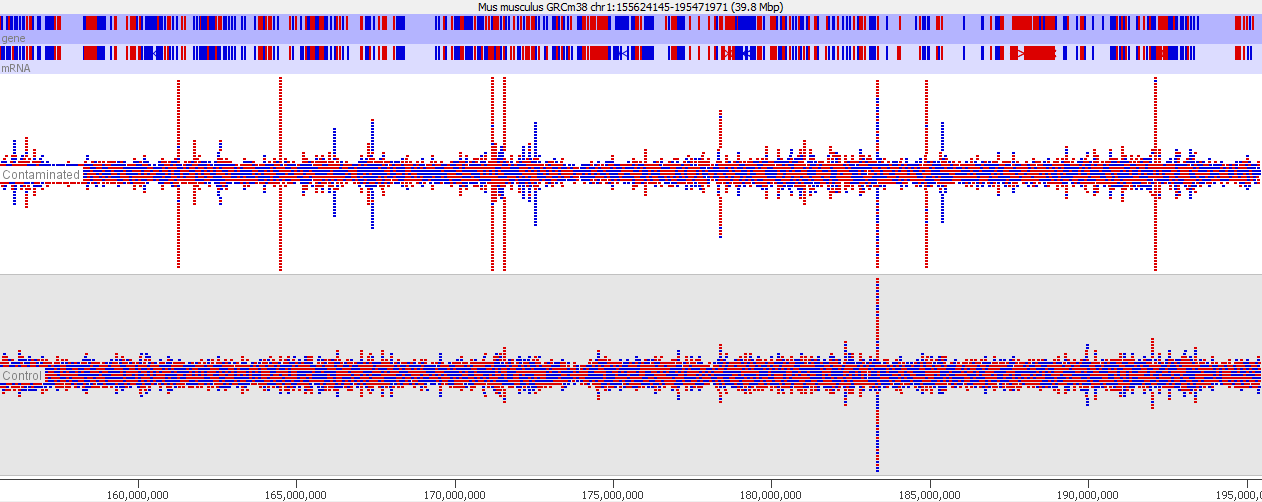
On the face of it it looks like the top sample has a number of extra peaks which would appear to be novel binding events. A closer examination however makes these novel peaks appear somewhat suspicious. In particular you can see that the novel peaks are directional (red = top strand, blue = bottom strand), whereas ChIP peaks would normally not be expected to exhibit any directionality.
If we look more closely at one of the peaks then things get more suspicious.
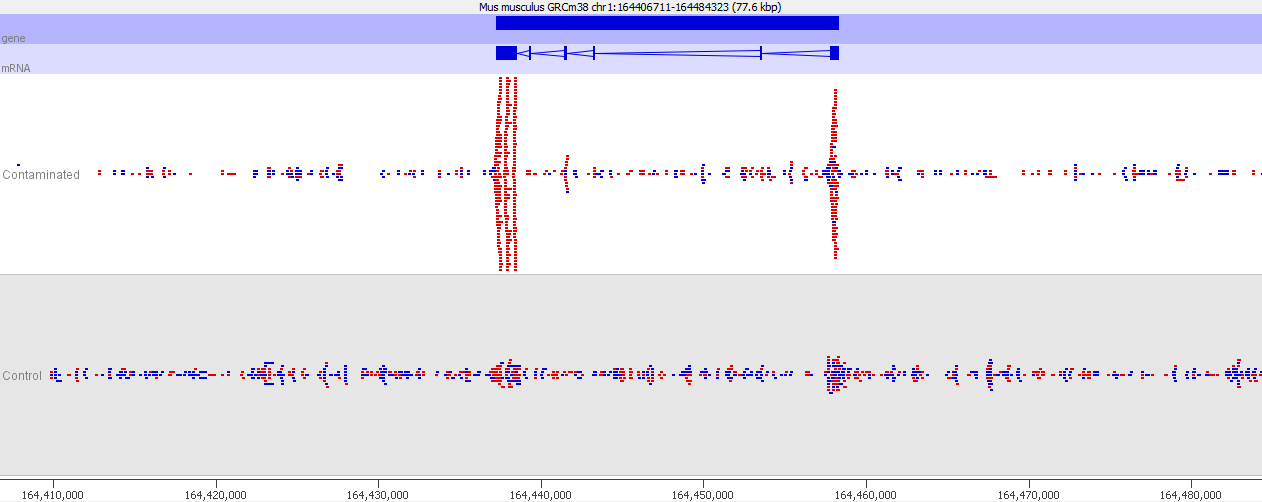
Now we look more closely it becomes very obvious that this is an issue of contamination. The extra peaks sit within the exons of transcripts and their directionality is always opposite to the direction of the feature. Our suspicion therefore would be that this ChIP sample has become contaminated with a reverse strand specific RNA-Seq library. Looking at the sequencer annotation we find that this is exactly what has happened – the user had mixed ChIP and RNA-Seq samples in the same lane and even though the libraries hadn’t seen each other until they were mixed within the flowcell, we still see this mis-annotation of reads.
The level of cross contamination seen here is very high compared to what you’d normally expect, but in this case it was exacerbated by a secondary effect – this was a highly duplicated library, so the user had deduplicated their data. The deduplication had removed a large proportion of the ChIP library, but the contaminating RNA-Seq data was not duplicated and wasn’t affected by the deduplication – amplifying the effect of the contamination. The same sort of effect will happen in other libraries though but may not be as obviously visible.
Mitigation
There is no way to completely prevent mis-annotation of barcoded reads in a sample. There are enough cases where you see a perfect, but incorrect, barcode that there’s nothing you can do to remove these. This may be an optical issue (mismatching the reads from barcodes and inserts), or some kind of internal recombination the flowcell, but really the exact source doesn’t matter for the purposes of this article.
The mitigation here is to make sure that the way you mix your libraries together will mean that it won’t matter if a very small percent of reads leak from one library to another. Generally this means keeping the same library type in the same lane.
If you have two similar libraries (genomic libraries for example) in the same lane and a tiny proportion of crossover occurs then since the overall distribution of reads between the two libraries is likely to be similar the proportional effect on any given position within the libraries will be negligible. However if your libraries have strong biases in their composition (RNA-Seq for example) then a random selection of reads from these will not be randomly distributed over the genome. A highly expressed gene in RNA-Seq could potentially have millions of reads over it, so that tens or hundreds of these reads could end up cross contaminating other libraries, making it look like something interesting was happening at this position.
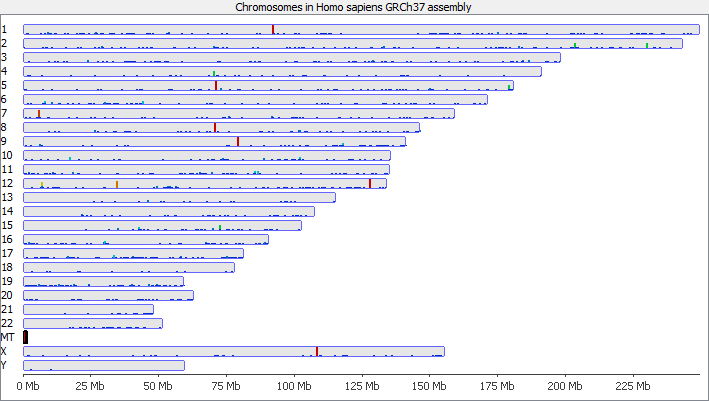
To illustrate this I took a pretty normal RNA-Seq library with ~20million reads in it and randomly select 2000 reads from it (that’s around 0.01% of the data – easily within the range of mis-annotated reads). You can see that the reads are not uniformly distributed over the whole genome but preferentially fall within a few regions which relate to the most highly expressed genes.
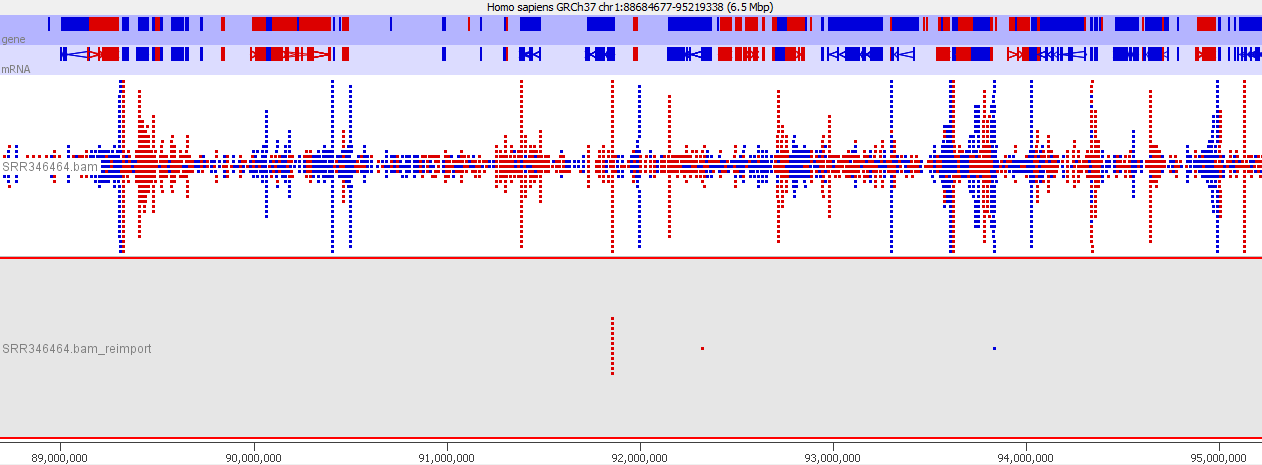
Looking in more detail at a single locus you can see that although the absolute number of reads is not enormous, if these were overlaid on a flat background they would appear to be a significant local enrichment. For enrichment seeking library types such as ChIP this could easily be misinterpreted, and if you were looking for rare events in a SNP detection or something sensitive such as a Hi-C experiment then this could appear to be a hugely enriched signal.
Lessons Learnt
Sequencers aren’t perfectly clean in their ability to separate barcode mixes put into them. You can minimise the effect of cross-contamination by only mixing samples from within the same experiment, but even then you might want to consider whether your results could be biased by the addition of a small number of randomly selected reads from another library in the same lane.
10 thoughts on "Mixing sample types in a flowcell lane generates cross contamination artefacts"
Yannick Wurm
Perhaps you may want to rename the post: “Mixing sample types in a flowcell lane facilitates detection of cross contamination artefacts”.
I think too many people are unaware of how frequently this kind of thing happens. I am much more comfortable seeing (and thus quantifying) what I know is contamination than suspecting that there might be some…
Simon Andrews
The problem is you can’t remove it accurately. You may be able to get some kind of measure for the overall rate of cross-contamination, but you won’t be able to accruately remove each individual read which comes from the other libraries.
Roy
How does one create the second last figure? Overlaying quantitative data on to chromosome view?
Phil Ewels
Hi Roy, all figures in this article are taken from SeqMonk. You can find it here: http://www.bioinformatics.babraham.ac.uk/projects/seqmonk/
Simon Andrews
Within SeqMonk, once you’ve quantitated your data you simply need to select a data store from the data view at the top left to have it’s data show up in the genome view.
James Hadfield
You say the mitigation is “[to mix libraries so that it won’t matter if a very small percent of reads leak from one library to another]”.However the current problems with PCR-free libraries on HiSeq 4000, X-Ten and probably NovaSeq are as bad as 1% contamination. Even if this is random and you’ve got 96 samples in the lane that’s still 0.01% which many circulating tumour groups are aiming for…including Illumina’s Grail.
A wet lab mitigation would be to use unique-at-both-ends barcodes as the swapping is happening randomly at either end of the library molecule, the chances of both ends being affected is REALLY low.
Long Wang
Illumina do need to work out a solution for this. This issue is pervasive in Illumina platforms. It could generate tens of thousands false variant calls in WGS data. Here is an article describe this issue: http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0120520.
The article termed this issue as “Sample Bleeding”. This issue was most possible due to clustering errors, or owing to the bridge amplification process (I get this answer from BGI). The barcodes would help less in any case.
Currently I found two phenomena of this might be of useful. 1) the contaminated reads was very few in amount, mostly resulted in low allele-fraction variants; 2) most frequently found in regions with ultra high coverage (higher chance to be mis-assigned to anther sample). So always be cautious with those low allele-fraction variants, especially in repetitive regions.
Raj Sasidharan
Interesting post! I found reference to this blog post from the following article on wired.com that talks about issues the authors at Stanford (Rahul Sinha and colleagues) found with HiSeq 4000, NovaSeq machines with ExAmp reagents and low starting material.
https://www.wired.com/2017/04/geneticists-fear-illuminas-sequencers-may-distort-results/
Link to Rahul Sinha’s preprint that came out in bioRxiv posted on April 9, 2017:
http://biorxiv.org/content/early/2017/04/09/125724
Ilumina says it has been aware of this issue and published a whitepaper on “Index hopping” or “Barcode hopping” as they call it on April 17, 2017:
https://www.illumina.com/content/dam/illumina-marketing/documents/products/whitepapers/index-hopping-white-paper-770-2017-004.pdf?linkId=36607862
Steven Wingett
Yes, the similar problem affecting patterned flow cells is an interesting topic. We hope to post something on this site shortly regarding this matter.
raul
Good article Simon! We had stumbled into this issue some years ago with the HiSeq 20/2500. I was given the task to find a informatic solution the problem. I noticed that doing the lane demultiplexing without mismatches in the barcodes (larger effect) plus doing a filtering of reads based on the index read average quality (smaller effect), did reduce the issue significantly. I wanted to write a short communication about it but wasn’t given a green light on this.
I believe the issues with the the HiSeq 4000 although similar, have different causes being that pattern flowcells use a different chemisty..
Comments are closed