Introduction
We generally think of contamination in samples coming from a completely external source such as a different sample or another species, but internal contamination is a real problem and can cause problems in the analysis of sequencing experiments. One of the most commonly seen internal contaminants is the presence of DNA in RNA-Seq samples. This type of contamination is very easy to miss and won’t generally trigger any errors in a standard RNA-Seq analysis pipeline, but can cause significant bias in the quantitated data.
Most RNA-Seq library preparation protocols include a DNAse treatment in the early stages to try to prevent the inclusion of any genomic material in the library. In practice it is not uncommon for the step to be ineffective and for DNA to make it through into the final library.
The Symptoms
Unless you specifically look for this type of contamination you will not generally see any specific symptoms from it. Mapping efficiency will generally not be too badly affected, and these reads will not trigger errors or warnings in any of the standard analysis pipelines. The inclusion of this data will however negatively impact the ability to correctly quantitate and normalise the data.
Diagnosis
DNA contamination is fairly easy to spot as long as you look for it. As soon as you visualise your reads against an annotated genome the presence of DNA is normally fairly apparent as a consistent background of reads over the whole genome which isn’t noticeable affected by gene boundaries. The reads should also have no directionality bias which makes them even easier to spot in directional libraries.
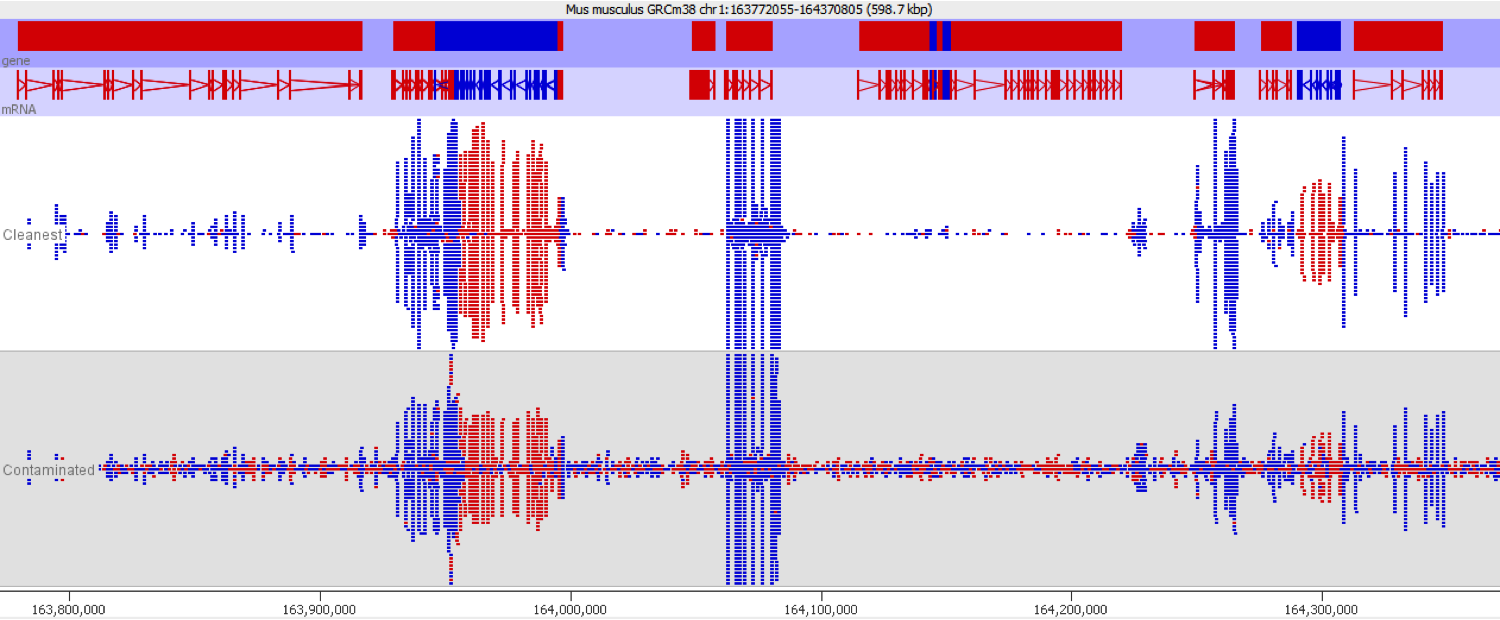
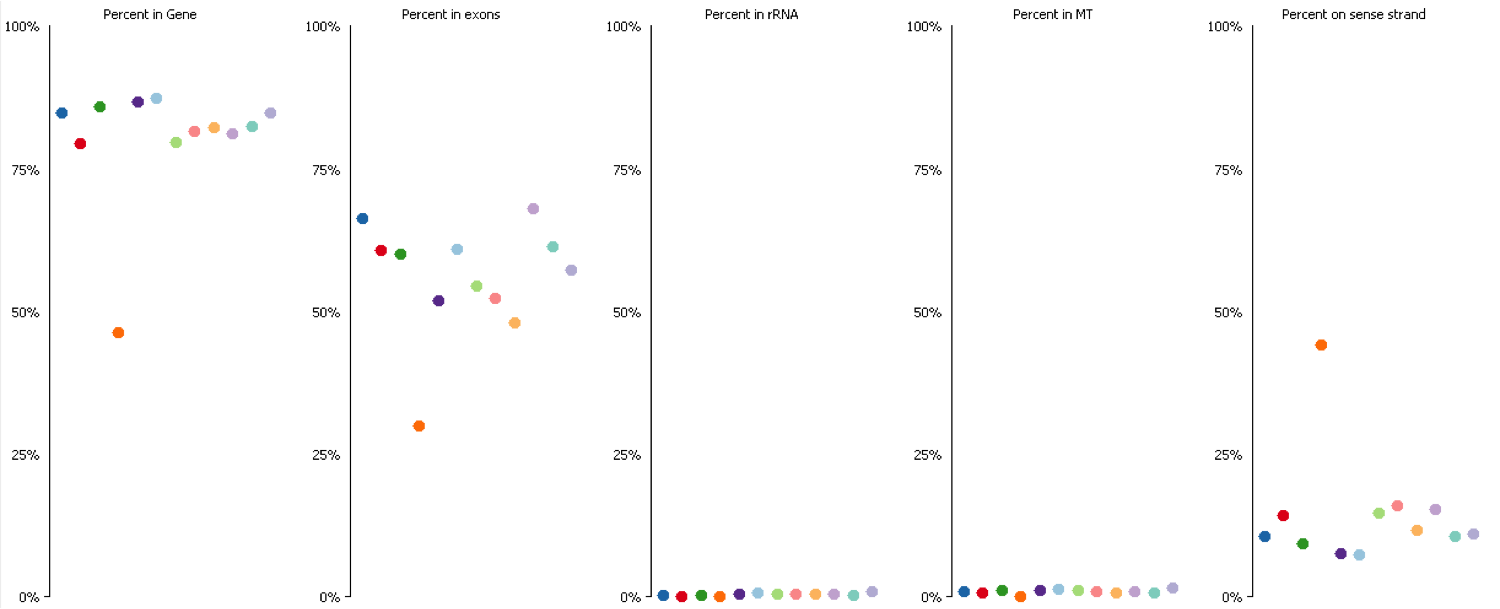
Since it isn’t always possible to visualise all datasets in large experiments you can also identify contamination by measuring some standard paramters such as the proportion of reads falling into genes, and the directionality of the reads.
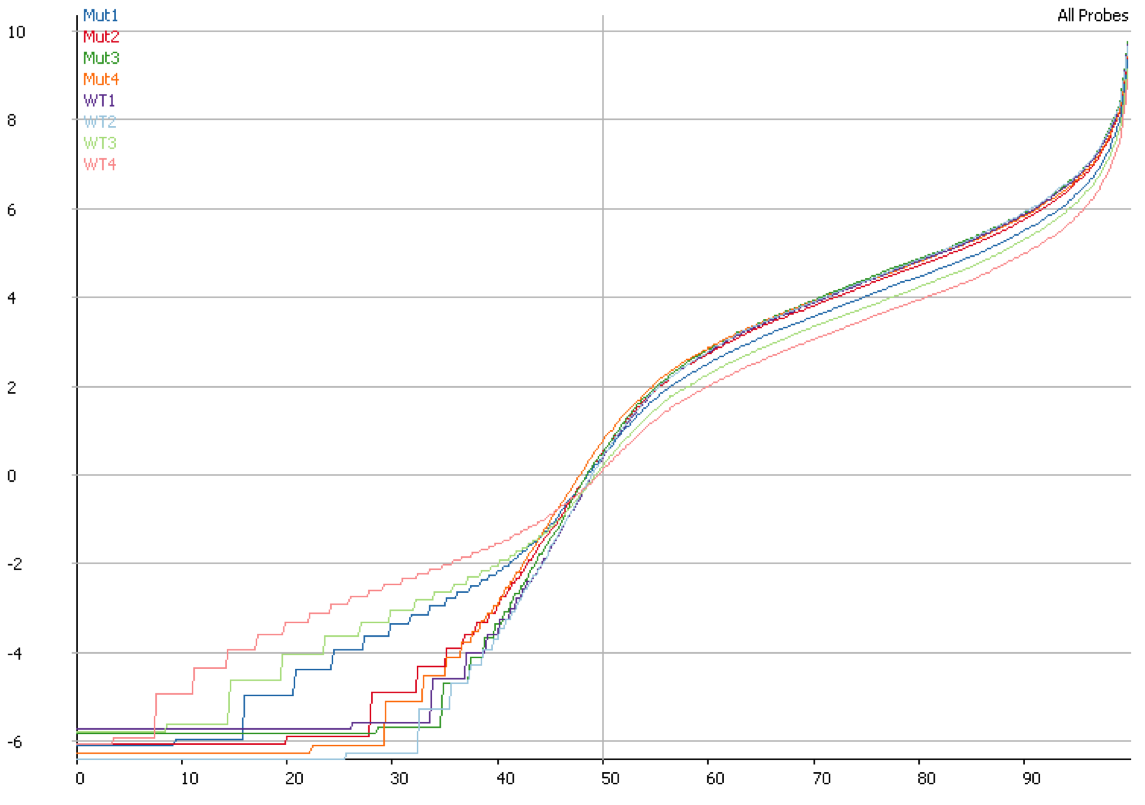
DNA contamination will also affect the quantitation of your data. If you compare the distributions of contaminated and non-contaminated samples then the contaminated samples will appear to have a loss of effective control on non-expressed genes which will all appear to be slightly expressed due to the random nature of the DNA derived reads. On more highly expressed genes the addition of DNA reads will have an insignificant effect, but the total number of reads involved can throw off global normalisation, and since the effect persists across a wide number of genes it can even throw off more robust normalisation schemes like size factor normalisation.
Mitigation
Once you have identified DNA contamination as an issue in your data it is possible to correct for it. In SeqMonk we use an option when we measure the median read density in intergenic regions over the genome on the assumption that this represents DNA contamination. This then allows us to estimate a per-transcript expected contamination level which can be subtracted from the observed count to try to get the DNA-free count. The plot below shows the quantitation for the same data shown in the previous plot after correcting for DNA contamination. It is clear that the overall distributions are much more closely matched after applying this correction.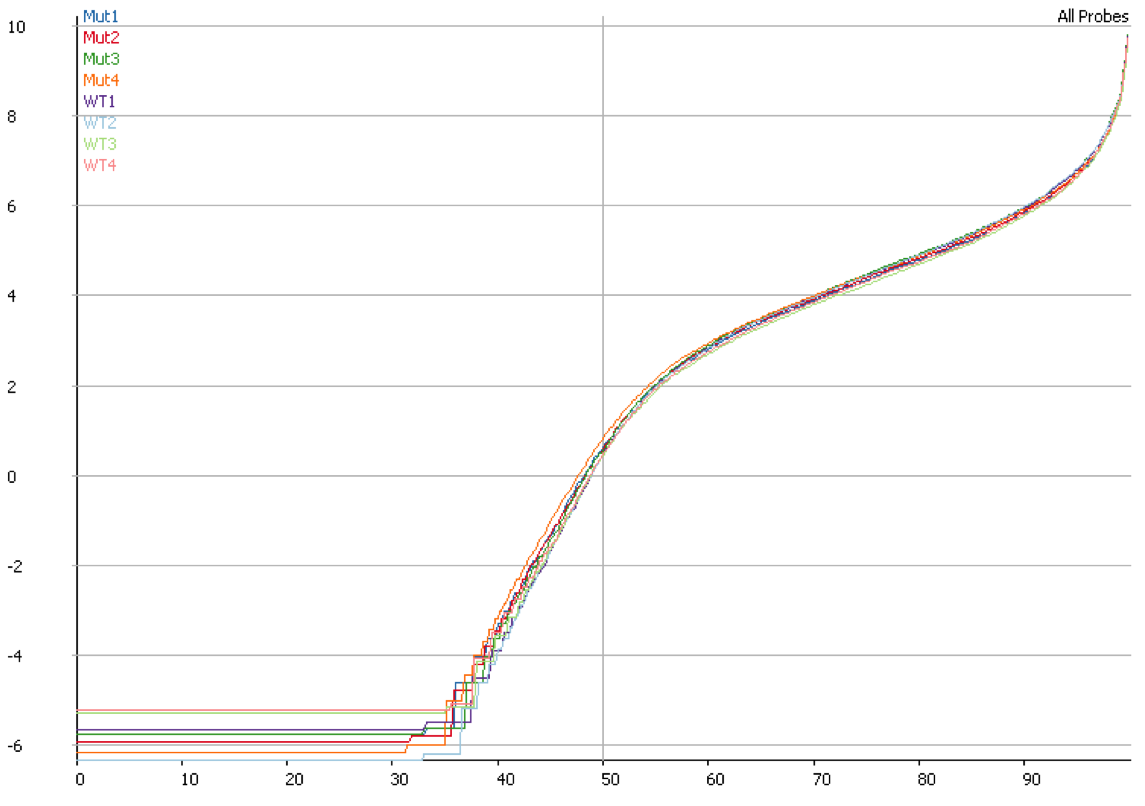
Prevention
This contamination can only be prevented at the library preparation stage through the rigorous use of DNAse treatment. Even then we regularly see instances of libraries with such contamination when such treatment was applied.
Lessons Learnt
Just because your pipeline runs smoothly doesn’t mean that there aren’t biases and artefacts in the data. Always find ways to view the key metrics for your raw and quantitated data.
Software
The plots in this article were all created with SeqMonk.
5 thoughts on "RNA-Seq samples can be contaminated with DNA"
Yatish B Patil
Hi,
Its a nice article.
I have one just basic question. Which .bam file I have to provide the one which is mapped to transcriptome or the one which is mapped to genome?. Because, in the RNASeq data quantitation pipeline, it quantitates for RNASeq.
Is that there is any option where we can get proportion of DNA contamination in the RNASeq data after removal of DNA contamination.
Please let me cleared this.
Simon Andrews
In general, when you map RNA-Seq data you map to the genome, even though you expect the majority of the reads will come from within the transcriptome. Mappers such as STAR or Hisat2 use this approach. In this case you figure out the potential level of genomic contamination by looking at the distribution of your mapped reads over the genome. Not all reads which fall outside the annotated transcripts will defintely be genomic (there could be problems with the annotation, or with mapping), but if non-transcript mapped reads are widespread then that’s a pretty convincing display that you’ve likely got genomic contamination present.
Lucas
Awesome! Thanks for that! Super helpful!
Serena Li
Thanks for sharing. I just have one project with DNA contamination problem spotted via RNA gel. One option is to re-purify the RNA samples. one earlier observation from ribosomal RNA depletion was that some transcripts were differentially depleted with n round of ribosomal RNA depletion vs. n+1 round and hence I was concerned by another biases introduced particularly if some samples were re-purified and others were not. Any thoughts?
asj
Nice Article!!
Is there a way to remove the genomic reads bioinformatically?
Comments are closed