Introduction
Shortly after the initial PBAT publication (which was single-end data), people started to optimise the protocol for extremely low starting material. We quickly noticed that the mapping efficiency for paired-end experiments (4N random priming initially) was rather low. We’re aware of adapter contamination and poor quality basecall issues, and more recently learned about mispriming biases in PBAT, but still the alignment efficiencies we observed were sometimes much lower than what we would expect despite rigorous adapter and quality trimming (we are talking about anything between ~30-50% for 2x100bp libraries).
The Symptoms
We then sought to identify why the mapping efficiencies were so low by hard-trimming single-end reads by a further 10, 20, 30 etc. basepairs from their 3′ end after adapter/quality trimming and removing the first biased 4bp (see here) with Trim Galore. We found that shortening the reads more and more resulted in ever increasing alignment rates despite the fact that really short bisulfite reads are significantly more difficult to align due to increasing the chances of multimapping reads because of the reduced search alphabet during bisulfite mapping (only G, A and T). This was not limited to our home-brew PBAT protocol but was also true for the originally published (single-end) data.
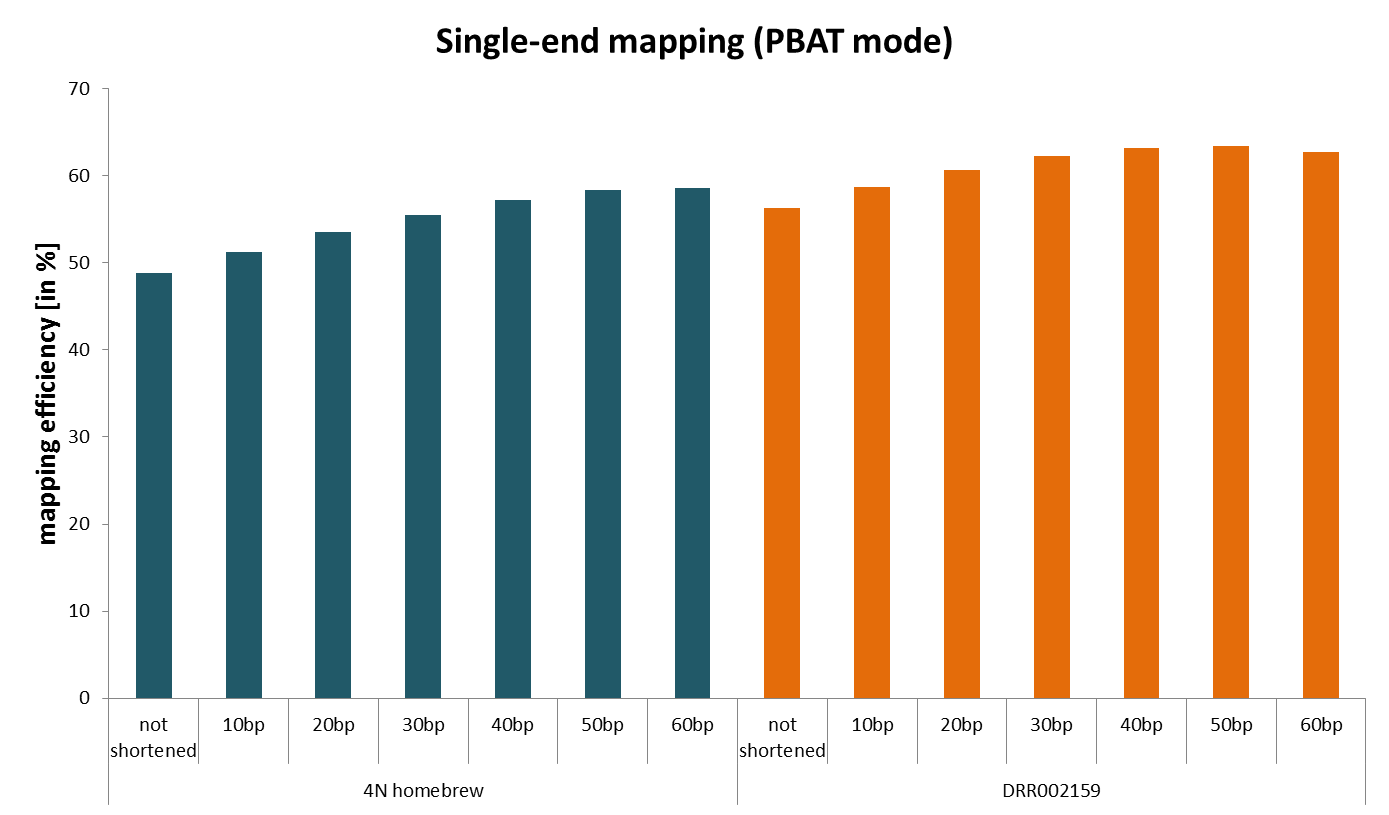
This indicated that something must have been present in the reads that prevented them from mapping efficiently.
Diagnosis
We reasoned that the sequenced fragments might be crossovers of different genomic sequences that were somehow generated during the two steps of random priming and strand extension. To test whether such chimeric reads really existed we aligned Read 1 and Read 2 of our paired-end library individually and teamed the aligned reads up by their read ID in a post-processing step.
The data was then imported into SeqMonk as a Hi-C input BAM file so that we could keep track to of what partner reads were doing. Shown below is a quantitation of all partner reads where the first end aligned to chromosome 1 (red = high number of partner reads, blue = low number of partner reads). This showed that the majority of Read 2 partners also aligned to chromosome 1, presumably most as valid paired-end alignments.
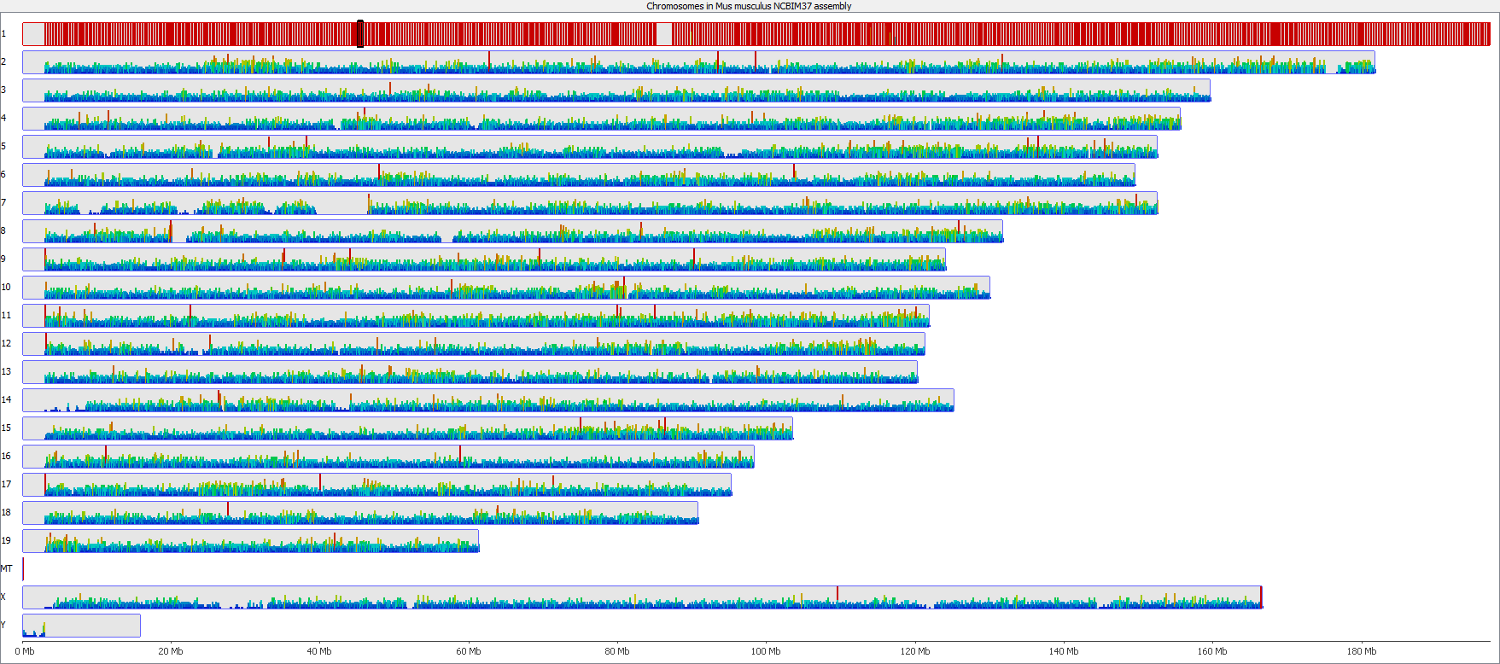
Strikingly however this also showed that there was an appreciable fraction of trans-hits, i.e. reads with Read 1 on chromosome 1 and Read 2 on a different chromosome. Even though there were a few hotspots (red bars) the interacting reads appeared to be spread out more or less evenly over the entire genome, so it was not just say a single type of repetitive element that preferentially formed read chimaeras. The number of trans-reads in the sample examined accounted for nearly 30% of all read pairs, so this is can be substantial problem.
Edit March 2019 for single-cell bisulfite sequencing (scBS-seq):
A recent article in Bioinformatics (https://www.ncbi.nlm.nih.gov/pubmed/30859188) also demonstrated in that chimeric reads are also the main problem for the low mapping efficiency in scBS-seq. In their article, Wu and colleagues demonstrate that the post-bisulfite based library construction protocol leads to a substantial amount of chimeric molecules as a result of recombination of genomic proximal sequences with ‘microhomology regions (MR)’. As a means to combat this problem the authors suggest a method that uses local alignments of reads that do not align in a traditional manner, and in addition remove MR sequences from these local alignments as they can introduce noise into the methylation data.
Mitigation
Such chimeric “Hi-C like bisulfite reads” deliberately do not produce valid (i.e. concordant) paired-end alignments with Bismark. To rescue as much data from a paired-end PBAT library with low mapping efficiency as possible we sometimes perform the following method (affectionately termed “Dirty Harry” because it is not the most straight forward or cleanest approach):
- Paired-end alignments (
--pbat) to start while writing out the unmapped R1 and R2 reads using the option--unmapped. Properly aligned PE reads should be methylation extracted while counting overlapping reads only once (which is the default). Also mind 5′ trimming mentioned in this post. - unmapped R1 is then mapped in single-end mode (
--pbat) - unmapped R2 is then mapped in single-end mode (in default = directional mode).
Single-end aligned R1 and R2 can then be methylation extracted normally as they should in theory map to different places in the genome anyway so don’t require attention to overlapping reads. Finally, the methylation calls from the PE and SE alignments can merged together before proceeding to the bismark2bedGraph or further downstream steps.
Edit March 2019: Please also see above the suggested mitigation approach for scBS-seq data using local alignments.
Prevention
Unfortunately the generation of chimeric reads seems to be inherent to the PBAT library preparation protocol and as such it is not very likely to just go away unless the random priming and extension steps could be optimised somehow. Even though a little cumbersome we found that using the PE alignments first and SE alignments afterwards approach achieves overall alignment rates that are almost in the range observed for whole genome shotgun BS-Seq. As a side note, even though the EpiGnome kit from Epicentre (an Illumina company) uses a PBAT-type amplification protocol it doesn’t seem to be affected very much by chimeric reads which is probably owed to their proprietary way of performing the pulldown/amplification step (which also generates directional and not PBAT libraries…). (Edit: The TruSeq DNA Methylation Kit is now discontinued).
Lessons Learnt
In addition to standard factors that affect most types of libraries (adapter contamination and basecall issues) and mis-priming errors at the 5′ end of reads, PBAT libraries may contain a substantial number of chimeric reads that prevent paired-end reads from aligning as concordant read pairs.
Datasets
Some datasets processed with the method described here may be found here under the GEO accession GSE63417.
Software
Data processing of public or homebrew PBAT data was carried out using Trim Galore and Bismark. Teaming up paired-reads was achieved using a custom written script. Visualisation of “Hi-C like” bisulfite read pairs was done in SeqMonk.
Using local alignment to enhance single-cell bisulfite sequencing data efficiency. Wu P, Gao Y, Guo W, Zhu P., Bioinformatics. 2019 Feb 19. pii: btz125. doi: 10.1093/bioinformatics/btz125. PMID: 30859188