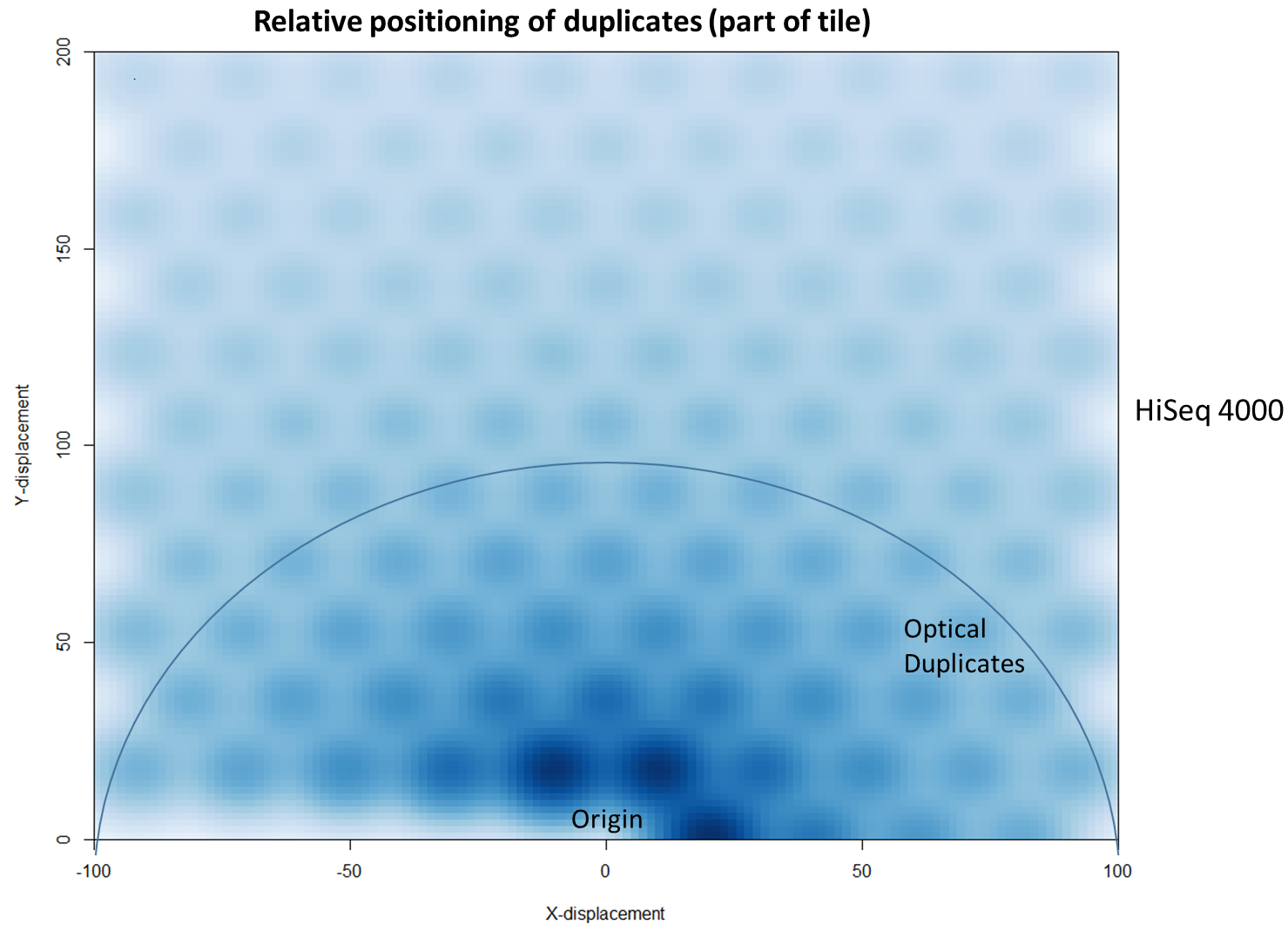
Illumina Patterned Flow Cells Generate Duplicated Sequences
The latest Illumina sequencers – such as the HiSeq X, HiSeq 3000 and HiSeq 4000 – use patterned flow cells to enable the discrimination between much more densely packed DNA clusters. While such technology substantially increases the number of reads generated per sequence run, this innovation may lead to an increased number of duplicates, thereby negating the improved yield and making subsequent data analysis potentially more difficult. Further investigation shows that these putative sequencing duplicates are generally in close two-dimensional proximity on a flow cell, which may provide an opportunity to develop bioinformatics solutions to identify and discard such artefacts.