Introduction
Next Generation Sequencing applications can interrogate a wide variety of biologically relevant molecules, events or modifications. Examples could be genome resequencing, RNA-Seq, ChIP-Seq or Bisulfite-Seq, to name but a few. The mapping efficiency (= the total number of reads that align) typically depends on several factors such as i) the technical quality of the reads, ii) the read length, iii) the alignment software used, iv) the absence of contaminants (such as other species or adapter contamination) and last but not least v) the quality of the reference genome assembly. Also the type of the experiment may have an influence, e.g. while RNA-Seq typically aligns back to the genome fairly well, techniques that tend to enrich repetitive sequences or Bisulfite-Sequencing (which only uses a 3-letter alphabet for mapping and often requires unique mapping) typically display somewhat lower mapping efficiencies.
Real life datasets are often not perfect however, they may contain poor basecall qualities, sequencing errors, insertions or deletions relative to the reference genome, unwanted sequences (e.g. contaminants) or read-through adapter contamination where the read length is longer than the sequenced fragment. While adapter/quality trimming and allowing for several mismatches and InDels allows to rescue most of these reads, a still fairly sizeable proportion of reads in the library may consist of sequences that are not present in the genome assembly and as a result cannot be placed at all.
The Symptoms
Despite the limitations of both reads and genome builds there are aligners out there claiming to have placed close to 100% of reads properly. This just sounds a bit to good to be true, so we tried to investigate this in a bit more detail…
Aligners reaching such high mapping efficiencies often perform so called “soft-clipping” of reads. This essentially means that portions of the read that do not match well to the reference genome on either side of the read are ignored for the alignment as such. This procedure carries a small penalty for each soft-clipped base, but it amounts to a significantly smaller alignment penalty than mismatching bases. Some programs even claim that this makes adapter trimming obsolete because as soon as the sequence does not align to the reference any more it will be soft-clipped.
Diagnosis
To get to the bottom of the differences between global, or end-to-end, mapping where the read has to align from the start to the end, and local mapping, where only a part of the read has to map and the ends may be soft-clipped, we decided to use several different library types that had undergone quality and adapter trimming using Trim Galore. This step should reveal the differences between end-to-end and local mapping without being affected by quality and/or adapter issues.
RNA-Seq
We started off by comparing some RNA datasets (single-end, 100bp, mouse genome GRCm38) using Hisat2 which in its current implementation (v2.0.3-beta) performs soft clipping (local, y-axis) by default (even though it is sort of undocumented), and the same dataset run using the option ‘–sp 1000,1000’ which effectively renders soft clipping impossible (end-to-end, EtE, x-axis).
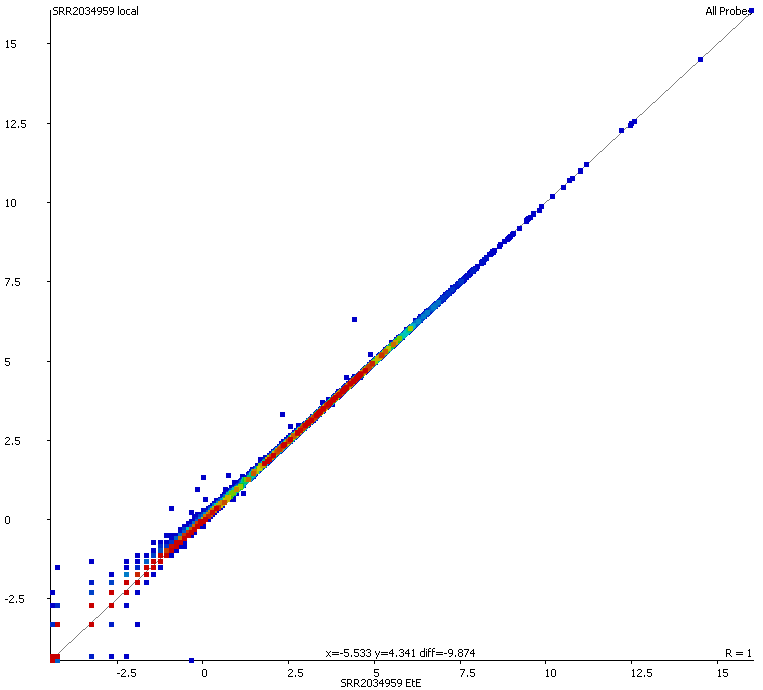
The data (quantified as log2 reads over protein coding transcripts) looked virtually identical between the two options, showing that for RNA-Seq data soft-clipping doesn’t seem to make a noticeable difference for downstream analysis. This is perhaps not surprising because RNA-Seq data represents rather well annotated and not very repetitive regions of the genome.
Genomic DNA
Next we looked at genomic DNA alignments, more specifically some randomly chosen ChIP-Seq or Input samples (either 50 or 100bp, single-end, mouse GRCm38 genome, again trimmed with Trim Galore). For the comparison we used Bowtie2 (v2.2.7) in end-to-end (EtE) or local (–local) mode.
The mapping statistics presented the following picture:
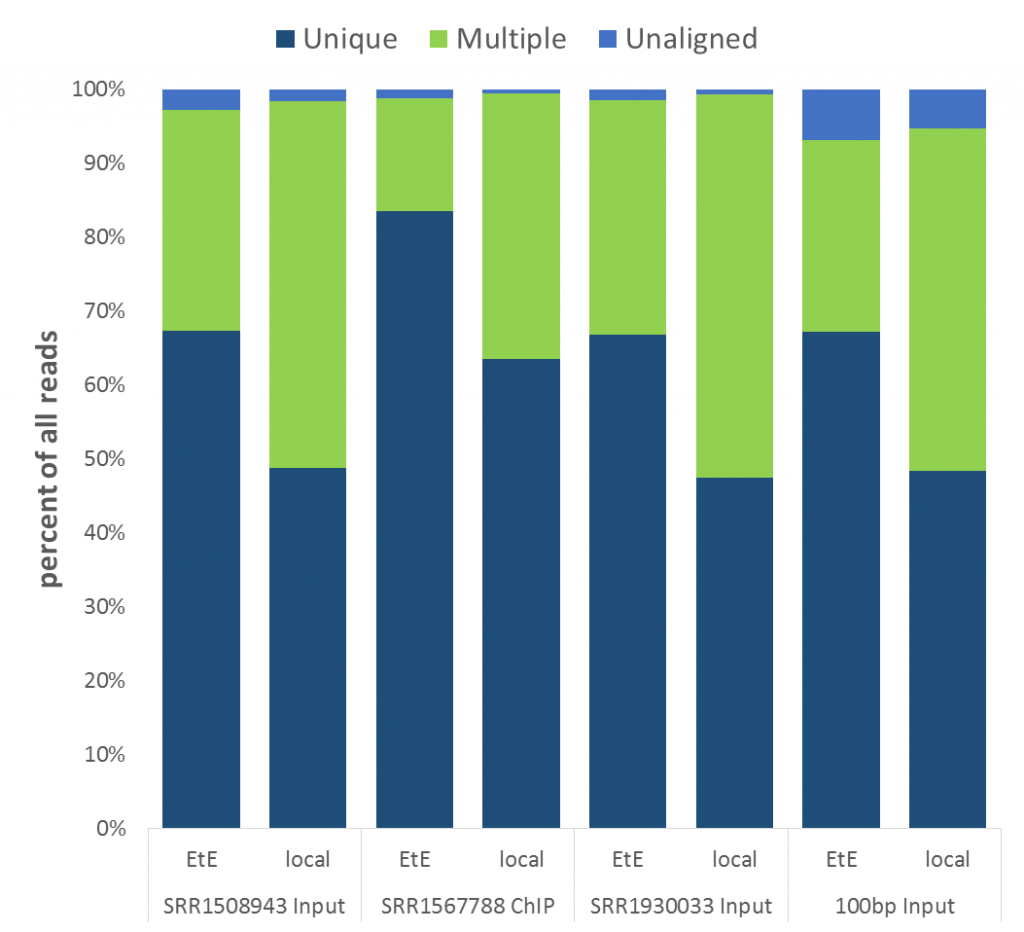
Soft-clipping was able to place more reads that were previously unaligned, but since the fraction of Unaligned reads was rather small to start with the effect didn’t look very dramatic. More striking was that in all data sets analysed the fraction of Unique reads was reduced while ambiguous alignments went up by roughly the same amount. In other words, it appeared as if reads that were previously aligned uniquely were soft-clipped to a shorter length so that they do now align to several places in the genome, i.e. repetitive sequences. As pointed out above we did not see this effect for the RNA-Seq data probably because it doesn’t contain as many repetitive sequences as applications using genomic DNA.
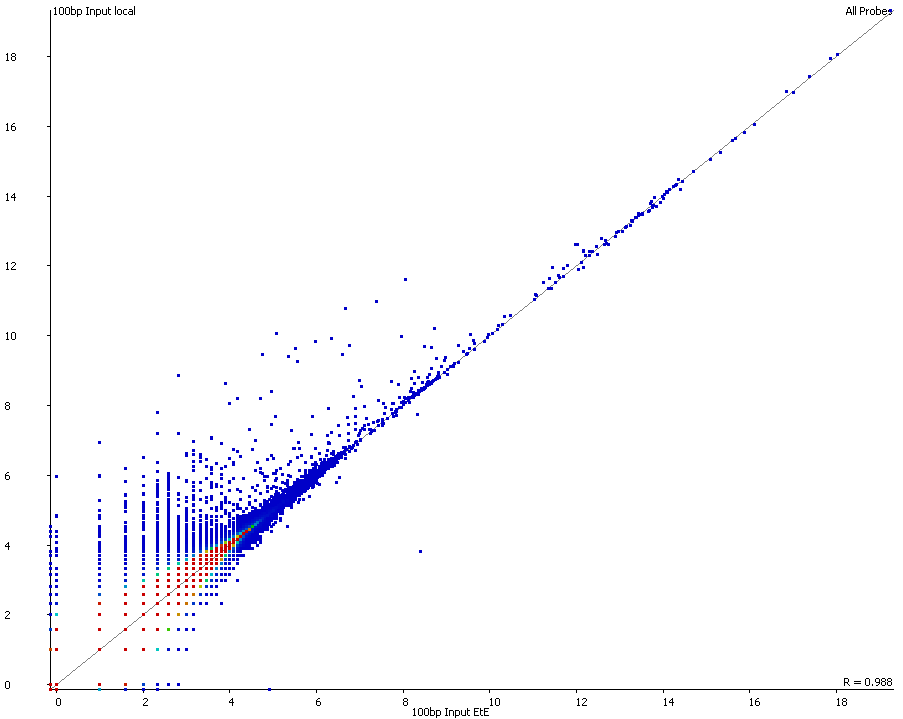
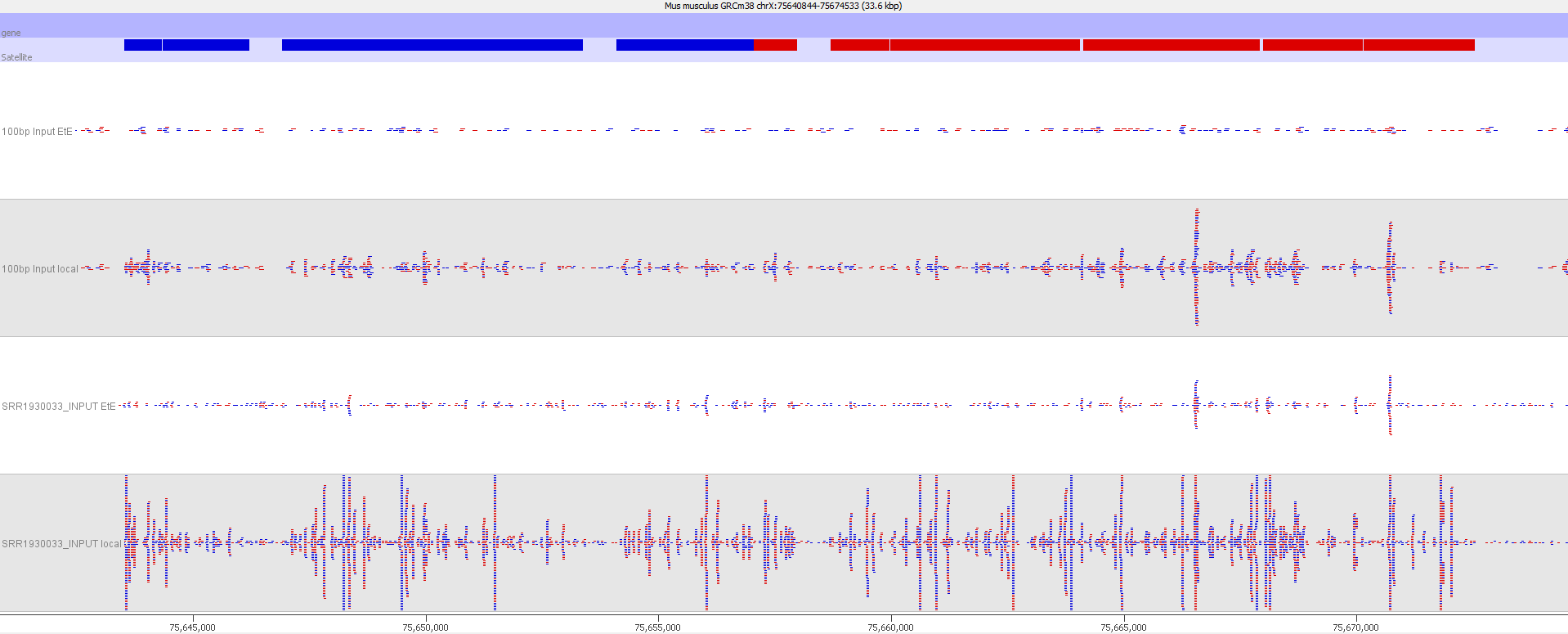
A scatter plot of end-to-end (x-axis) and local (y-axis) showed quite a number of regions with much increased read counts in local mapping (reads were quantified over 2kb genomic regions using log2 counts). When visualised on a genome-wide scale the regions with high read counts exclusively with local alignments accumulated suspiciously close to chromosome ends or other gaps in the genome assembly, suggestive of centromeric or telomeric repeats.
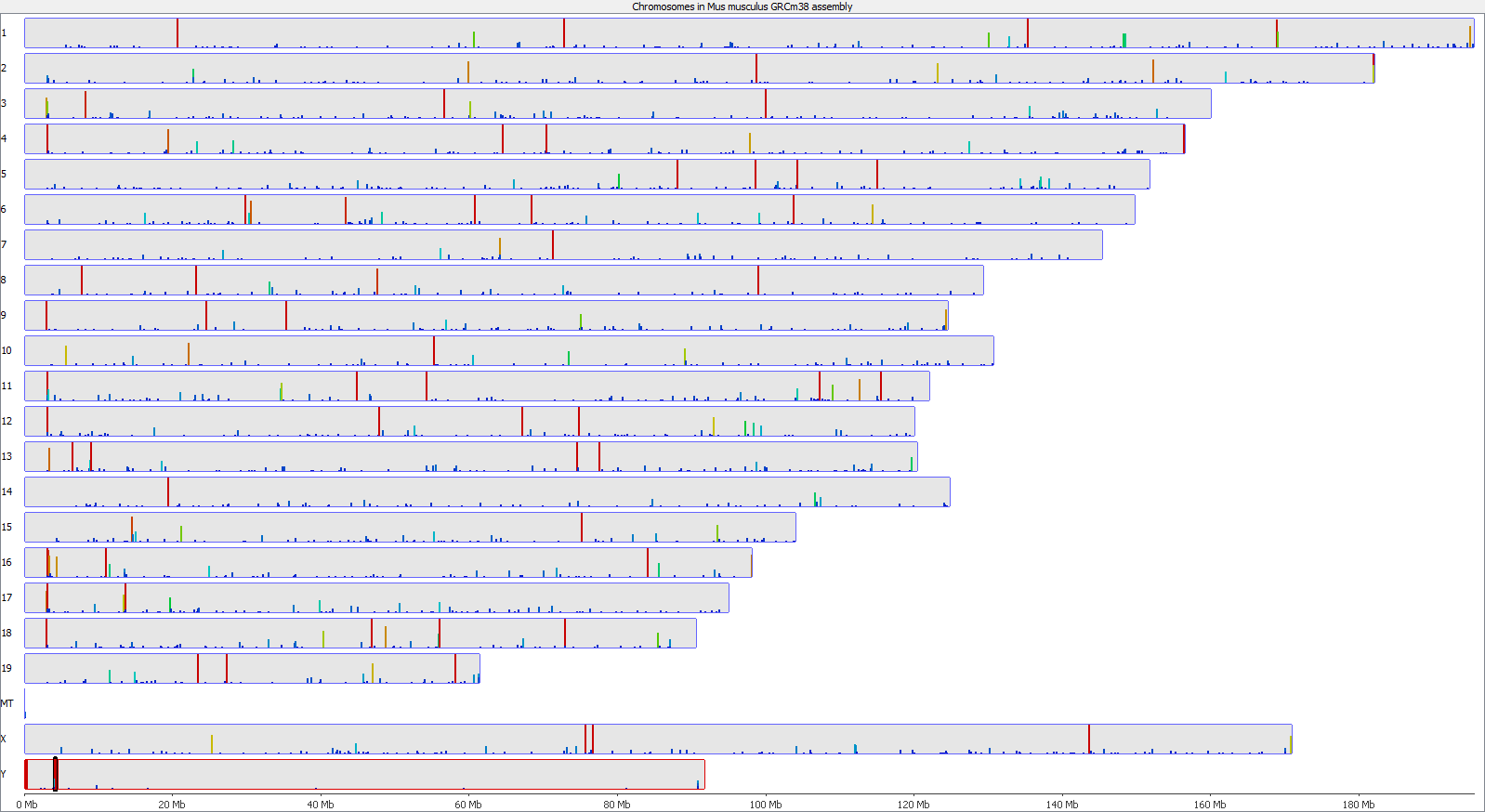
Indeed we found that a large proportion of regions (>80%) with high read counts overlapped annotated Satellite repeat regions (RepeatMasker), such as this one:
Bisulfite-Seq
Lastly we also wanted to take a look at Bisulfite-Seq (BS-Seq) because it is special for a few reasons:
- many aligners in-silico convert C to T in both reads and the reference genome during the mapping; this 3-letter alphabet approach makes it more difficult to place alignments uniquely
- most bisulfite aligners require unique alignments because it is not ‘safe’ to call methylation states if you can’t be sure where a read really came from
Because of these ‘features’ it is difficult to exceed alignment efficiencies of ~86% for human or ~78% for mouse for typical BS-Seq experiments (with say 100bp reads), yet soft-clipping still achieves alignment rates of 95-100% also in this setting. To look at differences between end-to-end mapping and soft clipping in a BS-Seq library we used 125bp human reads (trimmed with Trim Galore) in single-end mode against GRCh38 (plus decoys), and used Bismark (end-to-end) and bwa-meth (soft-clipping) as examples.
First of all it is good to see that the two methods generate virtually identical results for the vast majority of the genome as can be seen here (soft-clipping top, end-to-end bottom):

There are however also regions showing drastic differences in read depth (2kb tiled windows, log2 counts):
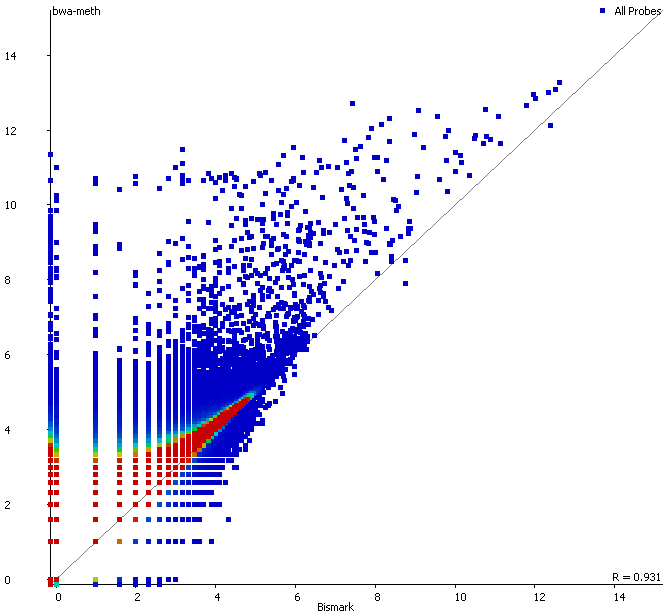
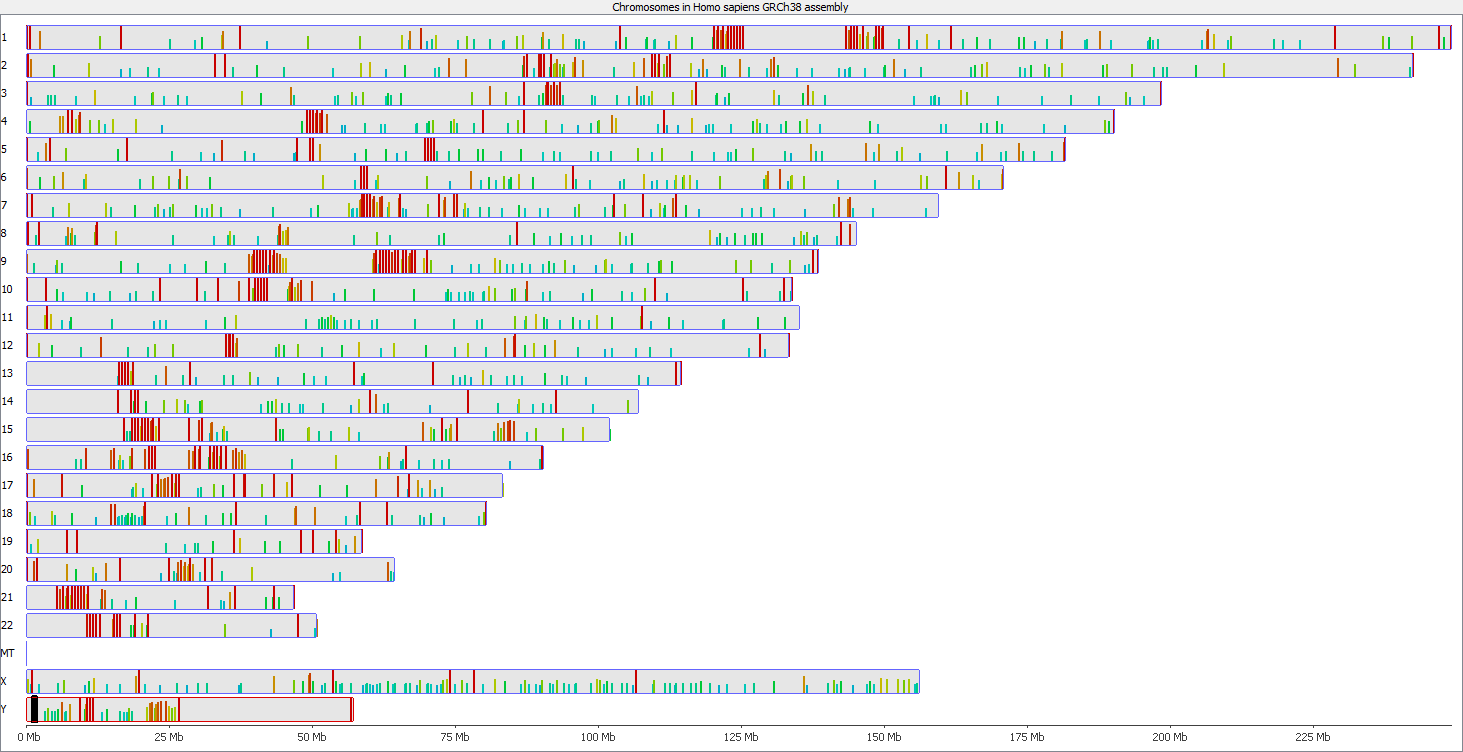
Regions with increased counts in the soft-clipped data do again cluster mainly towards ends of chromosomes or regions that have large gaps (or Ns) in the genome build.
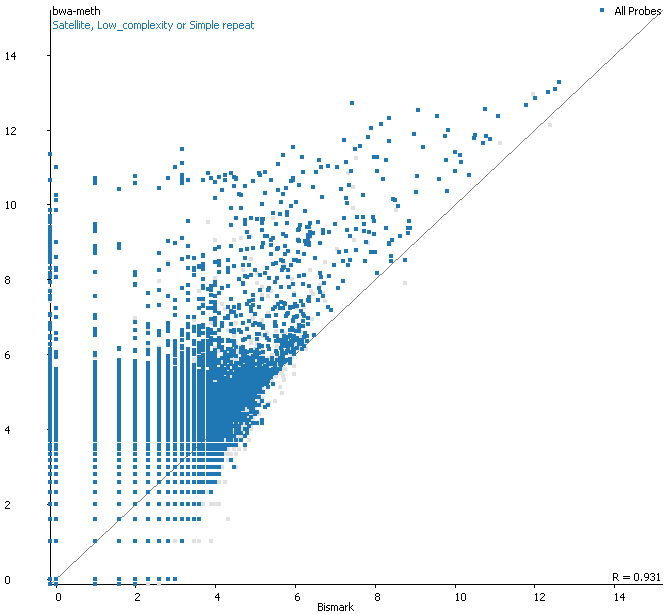
Filtering for overlaps with Satellite, Low Complexity or Simple Repeat regions for the human genome (RepeatMasker) explains nearly all of the coverage differences:
Here is a browser shot of one these regions illustrating the differences in read coverage (soft-clipping top, end-to-end bottom):
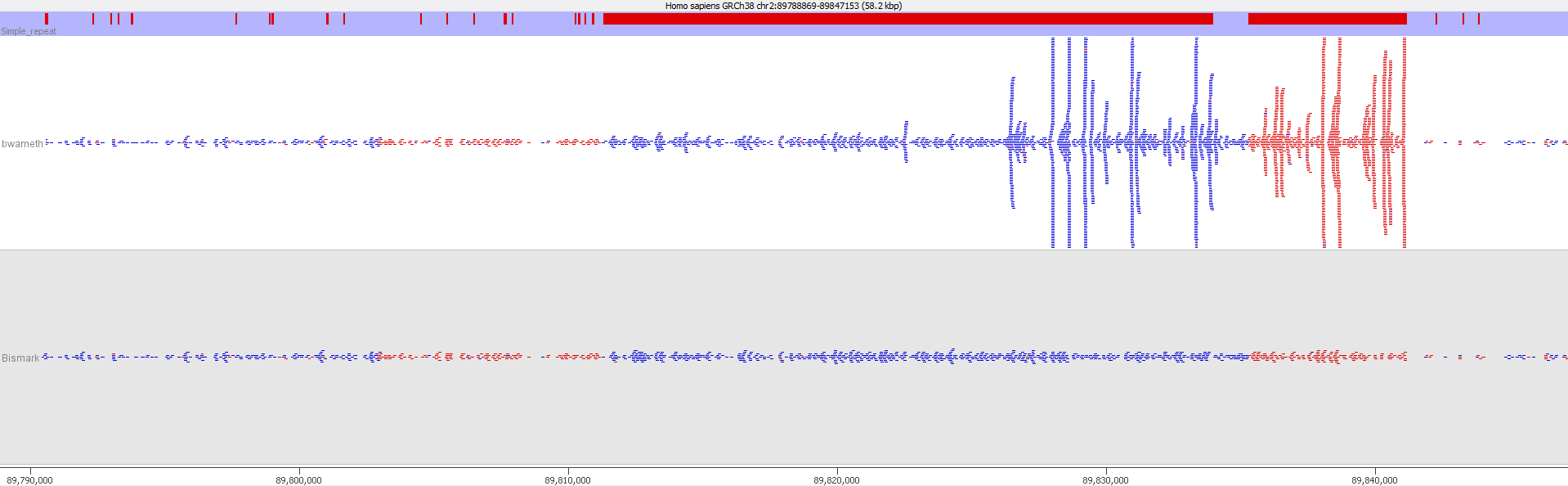
What happens in these cases is that reads which do not align anywhere in the genome (probably because they are not actually present in the genome build) are soft-clipped until a part of the read can be placed somewhere – unfortunately this newly ‘gained’ mapping position is not necessarily the correct one. While it might be informative to know that some part of a read resembles a certain type of repeat it is the wrong thing to place a read uniquely to a near-enough partial match of the sequence, and in case of bisulfite data then infer the region’s methylation state from such alignments.
Mitigation and Prevention
Luckily such hot-spots where soft-clipped reads get assigned incorrectly to repetitive regions in the genome can easily be spotted by their coverage, which tends to be hugely higher than for the rest of the genome. These reads could be removed by virtue of the abnormal coverage, but they are better handled by filtering on the read MAPQ values:
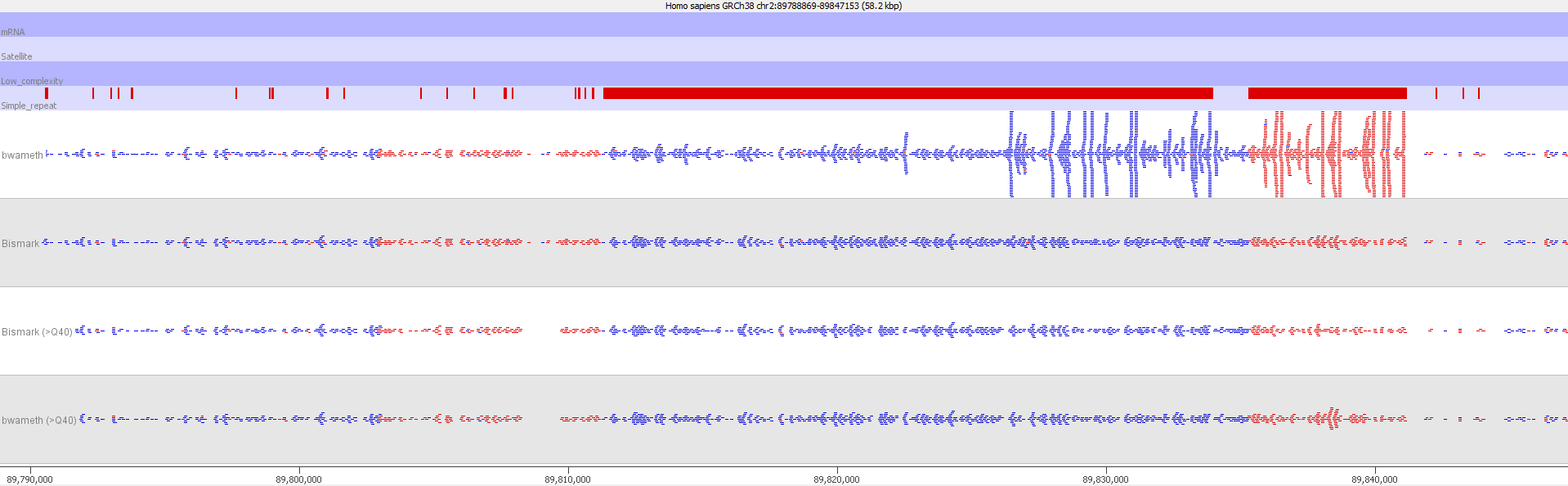
Filtering out reads with a MAPQ of <40 effectively removed all of the dodgy alignments, rendering the results virtually indistinguishable from end-to-end alignments again.
Lessons Learnt
The take home message from this is that soft-clipping of reads may force reads to align somewhere in the genome even if the location is not the true origin of reads, and this is most apparent for repetitive regions of the genome. This doesn’t mean that soft-clipping per-se is a bad thing, in some cases it might rescue some true alignments e.g. from technical problems such as the overcalling of Gs on the NextSeq platform (until it has been properly addressed by Illumina), or for the detection of structural variations in the genome.
For standard genomic alignments however we believe that it is important to understand the data at hand, know which kind of contamination may occur at the read ends (e.g. adapter sequence) and remove those and poor qualities accordingly. And one has to be aware that ‘magically’ increasing the mapping efficiency to close to 100% comes at the cost of potentially including a ton of incorrect alignments with all the side effects this may have on downstream analysis (e.g. correction, statistical power etc.)
2 thoughts on "Soft-clipping of reads may add potentially unwanted alignments to repetitive regions"
Chris Fields
Hi Felix, good article. One question re: Bisulfite-Seq and bwa-meth/bismark: what would you recommend re: filtering the alignments if they are paired-end?
Felix Krueger
Hi Chris, Sorry I was travelling in Yellowstone… I have to admit I’m not quite sure which filtering you are referring to for paired-end reads. Filtering out alignments with very low MAPQ scores would remove both read 1 and read 2. But note: reads aligned with Bismark use a very stringent default setting that would fairly quickly assign low MAPQ scores just based on mismatches even when the alignment is not necessarily non-unique. The filtering is more relevant if you use very relaxed parameters or allow soft-clipping (such as in bwameth).
Comments are closed