Introduction
Recently the popular technology and science magazine Wired ran a story discussing a problem with a new model of DNA sequencer. The article chronicled the experiences of a Stanford-based researcher who appeared to be making interesting findings while investigating blood stem cells. Initial optimism turned sour however, as it became clear that potentially significant results were the by-product of a problem with the sequencing technology, causing different samples to become mixed-up on the sequencer. While this was unfortunate, to say the least, for the scientist concerned (who reportedly lost a year’s worth of work), the implications of this technical problem set alarm bells ringing in the wider molecular biology and genomics communities.
The fact that a news outlet such as Wired covered this story pays testament to the potential impact of these findings. Although Wired has a technological focus, it is a publication intended to be of interest and intelligible to a general audience, and so it may be surprising to find a story about sequencing artefacts within its pages. The relevance of the story lay in the fact that the machine used was the latest technology offering from Illumina, the San Diego-based company seen as synonymous with sequencing by many in Life Sciences research. Indeed, the wide-scale use of these machines has left countless researchers worried that their work may be rendered invalid, with key findings in published work or current research programs simply being the result of Illumina machines muddling samples.
The Symptoms
The Stanford researchers deposited their findings on the bioRᵡiv preprint server. Their paper reported that they sequenced mouse single-cell RNA-Seq samples using a HiSeq 4000. As is common when working with single-cell data, they multiplexed the samples: a technique in which DNA molecules are labelled at either end with predefined barcodes, and then the DNA from different libraries are combined. This pool of samples and accompanying barcodes are then sequenced togehter, resulting FASTQ files may be “de-multiplexed” using specialist software, enabling researchers to deduce which read came from what library.
Suspicions were raised when investigating the spatial arrangement of expression patterns between samples on the 384-well plate used during sample preparation. Each well should have contained the cDNA from a single cell, and the samples should have been randomly distributed across the wells. Surprisingly, if a given gene was expressed at very high levels in a particular well, the signal was also detected – but at lower levels – in all or most wells in the same row and column, forming a characteristic cross-like expression pattern in the plot (see figure). Similar patterns could be observed when analysing other types of library e.g. ATAC-Seq. The researchers decided the pattern was most consistent with a being a technical artefact.
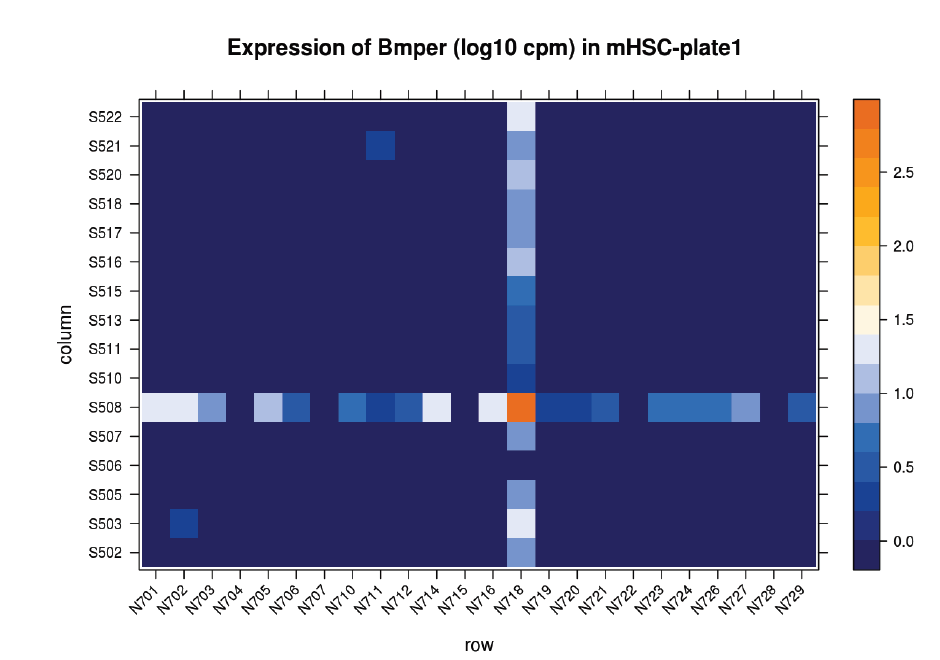
Each row and column correspond to different barcodes. The results show that Bmper is highly expressed in the single-cell sample barcoded with S508/N717. However, almost all the other samples with one of these barcodes also show elevated expression of Bmper. (Taken from bioRᵡiv.)
Diagnosis
The Stanford group re-sequenced their samples on another HiSeq 4000 and observed the same pattern, however the pattern was not observed when sequencing on a NextSeq 500. This was highly significant since the older model NextSeq 500 did not use patterned flowcells, suggesting the problem was specific to that patterned design. This patterned arrangement comprised billions of fixed-sized nanowells distributed uniformly across a sequencing flow cell, enabling the discrimination of separate DNA clusters at much higher densities than possible on previous generation machines.
The cruciform pattern of the mixed signal, which corresponded to the barcode allocation, suggest that one of the barcodes was being mis-identified. To investigate this further they performed an experiment in which wells were allocated either:
- cDNA / reagents (including index primers)
- Reagents (including index primers) only
- No cDNA / no reagents
Barcoded reads corresponding to wells containing neither reagents nor cDNA were very rare. In stark contrast, many reads (around 7% of the average for wells containing cDNA) were found in the wells missing only cDNA. Moreover, these reads mapped to the mouse genome (mm10) with around 80% efficiency. This suggests that the presence of index primers caused the mixing of the sample and consequently when handling multiplexed samples there will be a substantial mix of signals, referred to as “index hopping” or “barcode swapping”.
While the percentages are unsettling, other researchers have reported observing the same phenomenon, but at a significantly reduced level. James Hadfield, the Head of Genomics at CRUK Cambridge Institute, observed an index swapping at rate of around 0.1-0.2% which may not prove so problematic for most applications.
(In fact, a very low level of barcode swapping had been observed on the previous-generation non-patterned machines, but this level should only be a significant problem if mixing greatly differing samples e.g. combining RNA-Seq with ChIP-Seq samples.)
Prevention
Barcode swapping observed on the HiSeq 4000 was probably a result of the new design using patterned flowcells, and therefore probably should be expected to also occur on the HiSeq 3000 and HiSeq X Ten and the flagship NovaSeq machine. The use of patterned flowcells necessitates the need for a new type of sequencing chemistry, called exclusion amplification (ExAmp), replacing the cluster generation by bridge amplification used on the conventional non-patterned flow cell.
Technical details describing the new sequencing chemistry have not been made publicly available, but examination of the associated patents made it possible to understand the overall process and it is reasonable to assume therefore that index hopping occurs before cluster generation, since all the reagents needed for cluster generation are present in that reaction mix. Free index primers in that mix have the potential to prime library fragments and be extended by DNA polymerase. These molecules, incorrectly assigned barcodes, are free to generate DNA clusters. This is not the case for the conventional flowcell design in which free barcodes are washed away after the DNA is hybridised to the flowcell. Only after this step is DNA polymerase added and sequence extension can initiate.
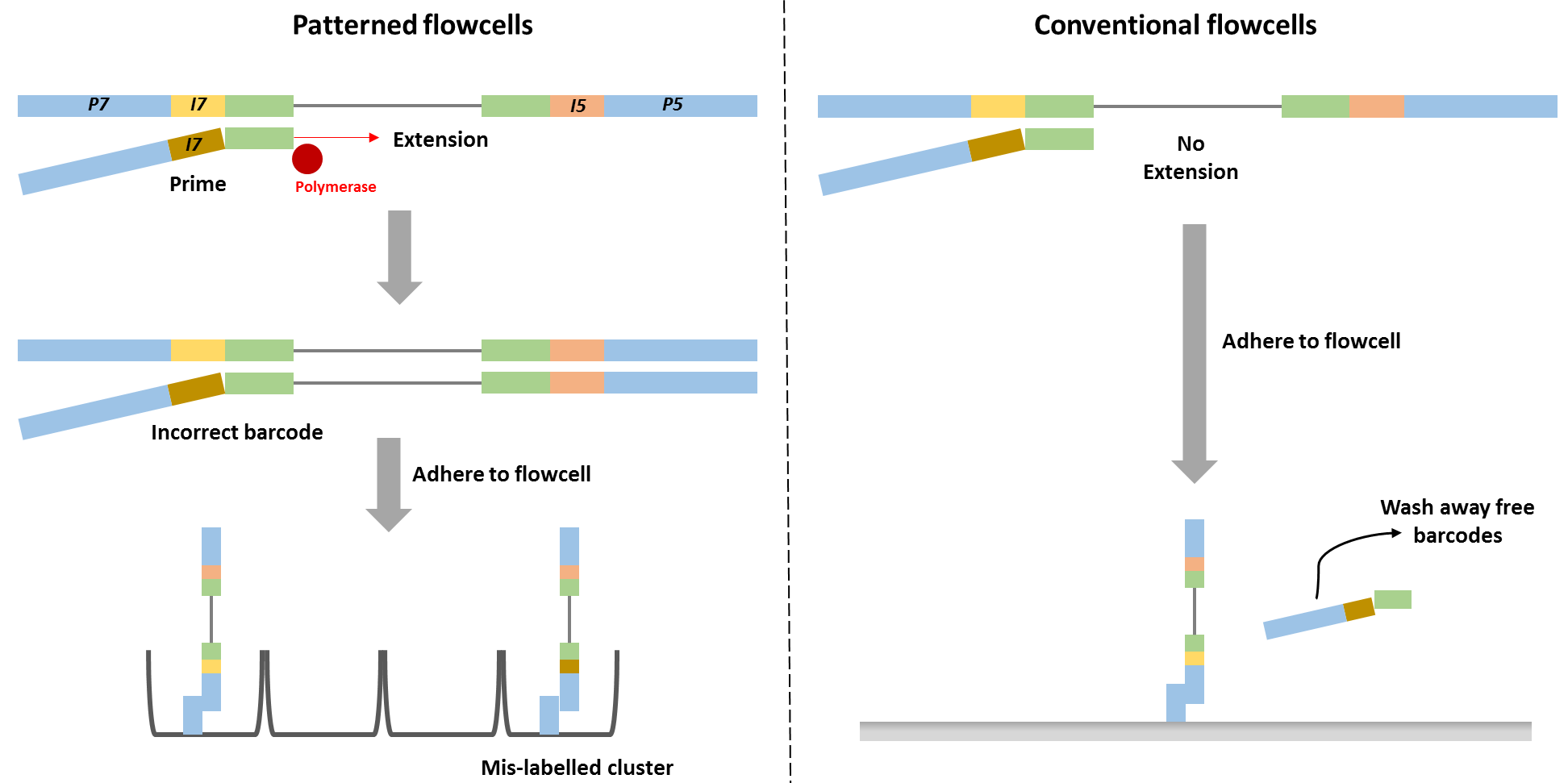
It is not clear at present how to implement a general purpose bioinformatics solution to remove such artefacts post-sequencing, and indeed experimental solutions may only be possible. To combat the problem, when working with a small number of samples, researchers should stop employing a single-barcode strategy, but instead should double-barcode samples. Following sequencing, reads should then be filtered to allow only those in which both barcodes are identical and have an expected sequence. Since double-barcode swapping should be a rare event, taking this step should greatly reduce sample misclassification.
When multiplexing many samples (already requiring double-barcoding, irrespective of the barcode hopping issue), Stanford researchers advised using barcode pairs in which each individual single barcode was used only once. Although that strategy reduced the number of available barcode combinations, it still allowed many samples to be run on the same lane. In addition, they proposed purification strategies to remove free primers from the library pool.
Illumina recently published a paper confirming the occurrence of index-hopping. They stated that with the proper clean-up of primers and implementing other experimental techniques, it should be possible to minimise barcode swapping to negligible levels for most applications. The report also advised only multiplexing similar conditions in a single lanes. A brain vs liver RNA-Seq example was given in which a transcript is present in liver at a high level, but not at all in the brain. Owing to index hopping that transcript may actually appear to be expressed at a low level in the brain. Illumina suggests that by only combining similar samples (i.e. either brain or liver) in a lane this problem will be prevented. This is far from an ideal solution however, since researchers will need to know in advance what to expect with regard to the expression profiles of their samples and furthermore this strategy would compound the problem of batch effects.
Lessons Learnt
Typically, our QC Fail articles report and analyse data generated in-house, at the Babraham Institute. For this article we have been unable to do this since our institute has not purchased a sequencer with the patterned flow cell design. Rather, we have provided a precis of the investigations of other teams.
Although we have been unable to actively contribute to the community’s knowledge by troubleshooting this problem directly, we, fortunately, have not been placed in the unenviable position of having to decide what sample may or may not have been affected by this problem and re-assessing results in light of this development. We feel therefore that researchers should exercise caution regarding becoming an early adopter of a technology that underpins all subsequently analyses, especially when such a new technology is intended only as an improvement over existing techniques and does not offer orders of magnitude fold improvement in terms of yield. The problem of barcode swapping, and other recent work which suggests increased duplicate generation on patterned flowcells, underlines the need for caution. Rather than being an early adopter, it may therefore be prudent to wait and see what are other researchers’ experiences, and hold-off using a new technology until the most salient technical artefacts have been discovered and effective workarounds, if needed, have been formulated.
One thought on "The latest Illumina sequencers muddle samples"
Justpassingby
I have questions regarding to how this hopping mess happen.
If I understand correctly, your hypothesis is that index primers from one sample anneal to other samples, are extended by polymerase, and this happen before cluster generation and only in patterned flowcell work-flow.
But where does polymerase come from? carryover by library itself? If yes, this is library issue and will be found on conventional flowcell too.(and also in single index library)
If this polymerase come from synthesis mix, does this mean DNA templates are mix with synthesis-mix before they are injected to flowcell? If yes, then this chimera extension happen during cluster generation not before.
And this index chimera will be also found in single index samples.
But there is no such things reported, right?
If there were single index chimera because of sequencing mechanism, not lousy post sequencing wash step, please let me know! To prevent index hooping is the very reason why I still stuck on single index system!
Btw illumina has already admitted conventional flowcell have index hopping issue too, through it is in a lower ratio.
Because both flowcell exhibit same problem, even if my understanding is wrong, your hypothesis doesn’t seem very convincing.
Comments are closed