Introduction
Library construction of standard directional BS-Seq samples is a multi-step process typically consisting of several consecutive steps:
- Sonication
- End-repair
- A-tailing
- Adapter ligation
- Bisulfite conversion
The end-repair step typically uses a mix of nucleotides to fill in fragments with a 5′ overhang which are frequently generated by sonication, leaving blunt ended fragments ready for the A-tailing procedure. This would not be apparent in other applications such as ChIP-Seq or RNA-Seq because the genomic sequence is simply regenerated in these fragments. The situation changes in BS-Seq applications though because it does not really interrogate the sequence itself but the methylation state of cytosine residues. The nucleotide-mix used in the lab typically uses a standard mix of the four nucleotides C, A, T and G for the fill-in reaction, whereby C is typically unmethylated for cost reason.
The Symptoms
This schematic drawing shows that cytosines located towards the 3′ end of reads may be filled in with unmethylated cytosines during the end-repair step. Since the fragments undergo bisulfite treatment after the adapters have been ligated on, these cytosines (marked in red) will be converted to T. Since Read 2 starts right from this end carrying the artificial bases they will be (incorrectly) read as unmethylated cytosines. If the sequenced fragments happen to be very short you might also see a read these filled-in positions at the 3′ end of Read 1, but this will be arguably quite rare, whereas the Read 2 bias definitely affects every single molecule with filled in cytosines.

It is actually pretty much impossible to spot the symptoms of this bias unless you really look for them as a post-alignment QC step (see below).
Diagnosis
The trouble with this kind of bias is that it only becomes apparent when you really look for it. To spot the bias one has to look at the average methylation levels across the entire length of the reads and plot out a methylation bias (or M-bias) graph:
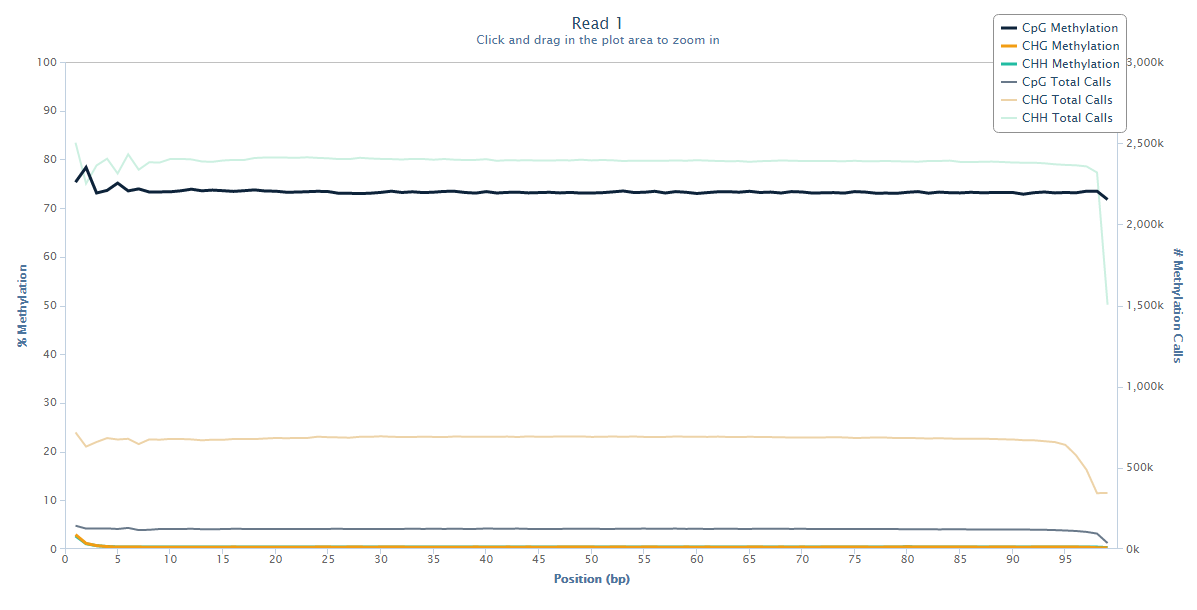
Read 1 looks fairly normal with a little bit of a ‘wobble’ at the 5-end but generally fairly even methylation levels across the entire read.
Read 2 however tells an entirely different story:
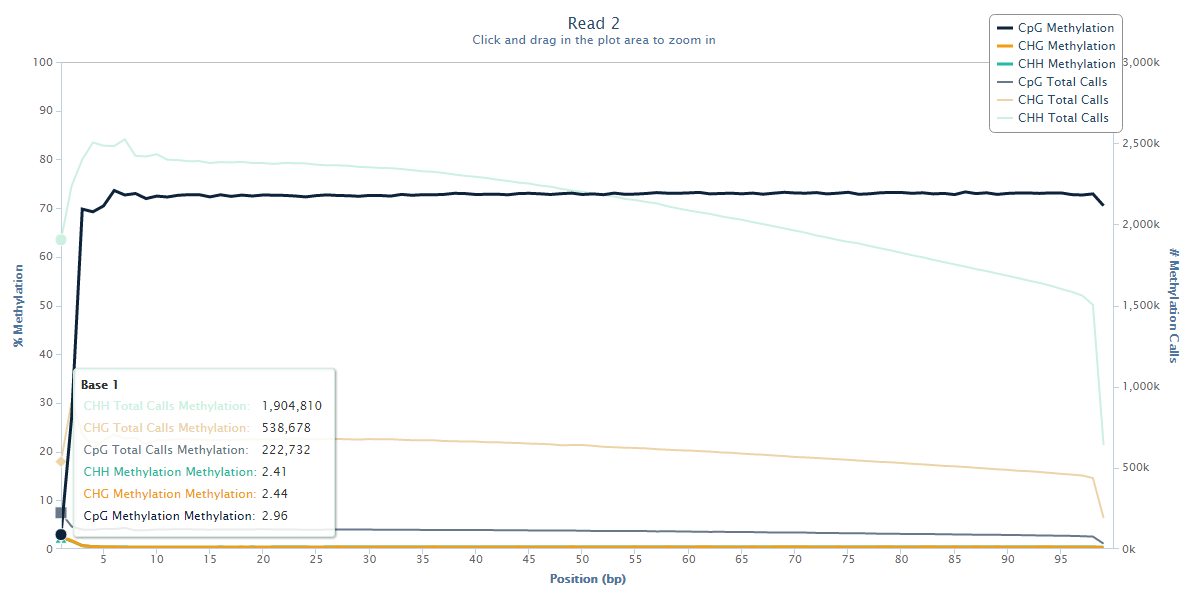
The methylation state of the first couple of bases or Read 2 drops from the average ~70% down to ~3%, and this steep drop can certainly be explained by the filled-in unmethylated cytosines. Since there is no reason to believe that there would be a biological role for lower methylation at the start of reads we have to assume that this low-methylation is artefactual, and thus leaving this untreated will introduce, in this case several hundred thousand, incorrect methylation calls (and thus noise) into the dataset.
Mitigation
Fortunately it is relatively easy to fix this problem by simply disregarding the biased positions when running the methylation extractor. Since this problem only affects Read 2 of paired-end libraries a typical command like this should fix the problem:
bismark_methylation_extractor --ignore_r2 2 --gzip sample1_bismark_bt2_pe.bam
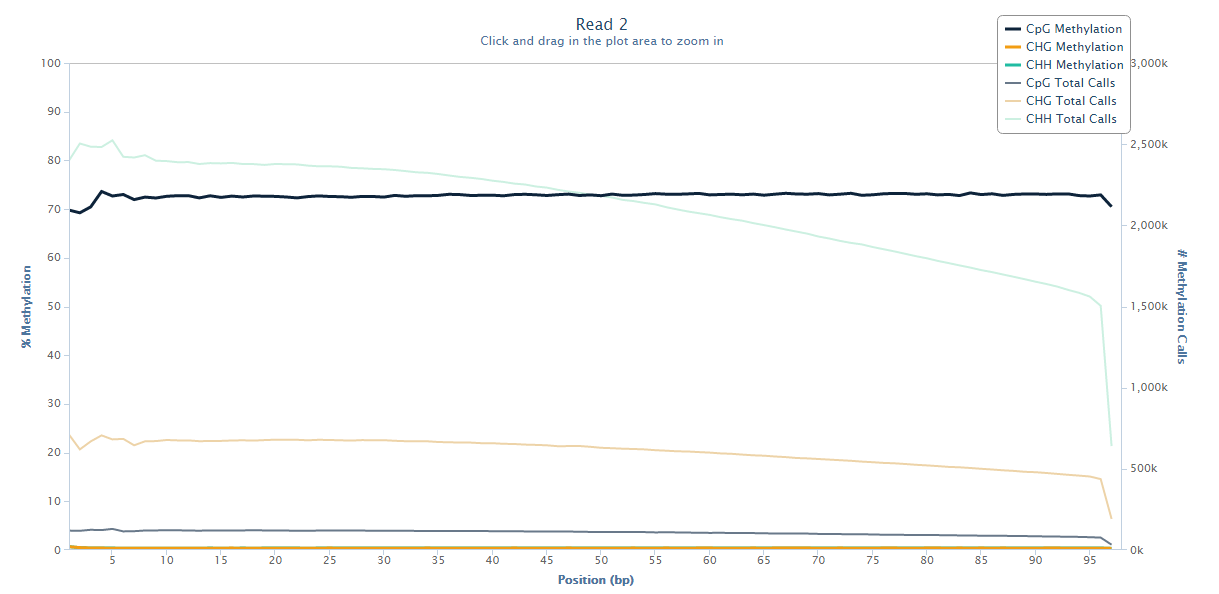
The same M-bias plot for Read 2 should then look like this:
Prevention
Since this is a common phenomenon with any standard directional library the option –ignore_r2 2 of the methylation extractor should be added to the default data processing pipeline for paired-end applications.
Lessons Learnt
Read 2 of paired-end BS-Seq libraries suffers from artificial hypomethylation at the start of Read 2 of paired-end reads. This will add spurious hypomethylated calls and should be removed from the dataset before proceeding to downstream analysis.
References
The M-bias plot was first described in this paper: Hansen et al., Genome Biology, 2012, 13:R83
Datasets
Any paired-end directional BS-Seq dataset will show this problem.
Software
To get to the stage where you can see the M-bias plot data was adapter and quality trimmed with Trim Galore and aligned to the mouse genome using Bismark. Methylation information was extracted using the bismark_methylation_extractor and reports shown in this article were generated using bismark2report.
3 thoughts on "Library end-repair reaction introduces methylation biases in paired-end (PE) Bisulfite-Seq applications"
Xinlei Hu
In Introduction part , it says that fragments with a 3′ overhang are filled in with nucleotides. But in The Symptoms part, it shows that the 3′ end of reads may be filled in with unmethylated cytosines—-I mean it’s 5′ overhang. According to Illumina’s support file of WGBS, the 3ʹ to 5ʹ exonuclease activity of the kit removes the 3ʹ overhangs and the polymerase activity fills in the 5ʹ overhangs. I get a little confused that which kind of overhang is filled in, or both overhangs are filled in .
Felix Krueger
You are absolutely right that it should read 5′ overhang in the introduction section, I have now updated the main article. Thanks for spotting this!
Roman Chernyatchik
Felix, thank you, good explanation. I have 2 additions.
1) You could notice a strong decrease in CHH (and less in CHG, CG) cytosines counts in R2 read close to 3′ end. It is because by default bismark methylation extractor ignores nucleotides from R1 and R2 overlap (–no_overlap option) in order not to count them twice. You are not supposed to fix this somehow because it is expected behavior. If you disable the default behavior (–include_overlap option) you will not see such bias.
2) For fragments close to read length you could notice R2 5′ bias at R1 3′ end (e.g. it is visible on the plots above). You could remove such bias using “–ignore_3prime” option.
P.S: ENCODE wgbs pipeline ignores 4 bp on both ends for R1, R2
Comments are closed