Introduction
Its is a common practice to re-use data deposited in one of the large public repositories. In some cases only highly processed quantitated data will be used, but often it is better to go back to lower level formats such as fastq files to run your analysis. In these cases we rely on the repository providing accurate data from which we can work but there are a number of ways in which this can fail.
One of the most problematic steps in the re-use of public data is extracting the fastq files from the database. The NCBI short read archive uses their own format, the SRA file, to store primary data. The SRA file is a composite file, much like a zip or tar file, which can contain multiple data sources within it, and NCBI provide a suite of tools, the sratools package to manipulate these files.
One of the most common operations on sra files is to extract the individual fastq files from the archive. For this process you use the fastq-dump program. If your data is paired end then you need to add the –split-files option to the program so that it writes out each read into a separate file. Omitting this will simply concatenate read 1 and read 2 in a single file, indicating that the internal representation of the read data in the file is simply a concatenation of the different reads.
Although this process works fine most of the time we have seen several cases where incorrect metadata in the repository leads to incorrect extraction of data from the file. Unfortunately this pretty much always still allows the extraction to complete without generating and errors or warnings so the issue may only be spotted later in an analysis pipeline.
The Symptoms
Although the failure here will probably be spotted in a larger pipeline by errors at the trimming / mapping stage it is first evident in the extracted fastq files. The obvious symptom is that the reads extracted will not have the expected length, and may contain odd characters (often a # symbol) in place of base calls.
For example if we have a look at sample SRR2133905. I can pull down the sra file from NCBI, and run fastq-dump successfully on it, but if I look at the files created then I see an odd split of data:
$ head -4 SRR2133898_Worker_3_Transcriptome_1.fastq SRR2133898_Worker_3_Transcriptome_2.fastq
==> SRR2133898_Worker_3_Transcriptome_1.fastq <== @SRR2133898_Worker_3_Transcriptome.1 HWI-ST412:281:D1D1BACXX:2:1101:1515:2212 length=98 TCGGATTCGTTCGATCCTTTTTTAAAGCTGACGATTTTTCTTTTTCTTTTCTTTTTCATCTAACGTGTTATTTTTGTTCTCTTTCTCTCTCTATCTCT +SRR2133898_Worker_3_Transcriptome.1 HWI-ST412:281:D1D1BACXX:2:1101:1515:2212 length=98 @<@DABDBFFHFH:CGCCHHCHHG?4?<3C3:)00BDEG*?DGGE88BGA)8)8@F@;;CCE####################################
==> SRR2133898_Worker_3_Transcriptome_2.fastq <== @SRR2133898_Worker_3_Transcriptome.1 HWI-ST412:281:D1D1BACXX:2:1101:1515:2212 length=1 C +SRR2133898_Worker_3_Transcriptome.1 HWI-ST412:281:D1D1BACXX:2:1101:1515:2212 length=1 #
You can see that read 1 has a length of 98bp and read 2 has a length of only 1bp, which doesn’t seem right. I can do the dump again, but without splitting files and get this:
$ head -4 SRR2133898_Worker_3_Transcriptome.fastq
@SRR2133898_Worker_3_Transcriptome.1 HWI-ST412:281:D1D1BACXX:2:1101:1515:2212 length=99 TCGGATTCGTTCGATCCTTTTTTAAAGCTGACGATTTTTCTTTTTCTTTTCTTTTTCATCTAACGTGTTATTTTTGTTCTCTTTCTCTCTCTATCTCTC +SRR2133898_Worker_3_Transcriptome.1 HWI-ST412:281:D1D1BACXX:2:1101:1515:2212 length=99 @<@DABDBFFHFH:CGCCHHCHHG?4?<3C3:)00BDEG*?DGGE88BGA)8)8@F@;;CCE#####################################
..which suggests that the sra file only contains data for 99 bases.
Diagnosis
To try to understand this I can go digging into the submission files. We can first look at the NCBI experiment details which show the following:
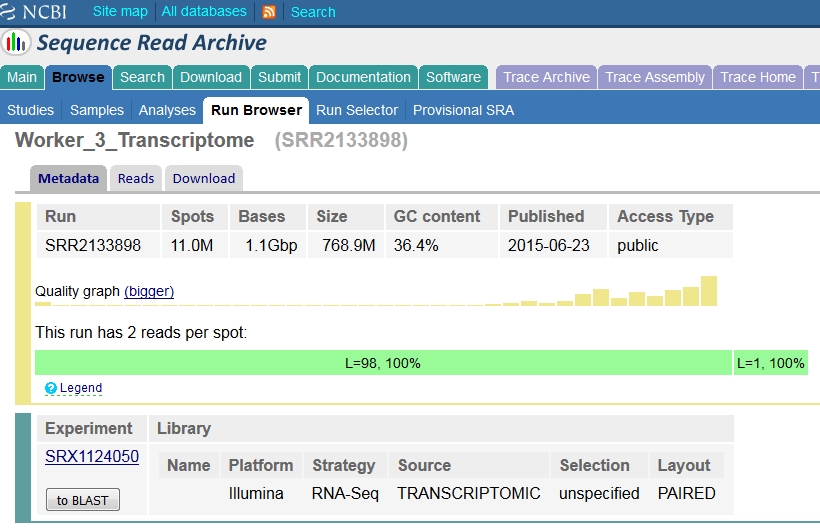
So the metadata here agrees with what we get from the extraction – a read of 98bp and a read of 1bp, but this doesn’t seem right. If we go to the metadata file at the ENA we see:
<SPOT_DESCRIPTOR>
<SPOT_DECODE_SPEC>
<SPOT_LENGTH>196</SPOT_LENGTH>
<READ_SPEC>
<READ_INDEX>0</READ_INDEX>
<READ_CLASS>Application Read</READ_CLASS>
<READ_TYPE>Forward</READ_TYPE>
<BASE_COORD>1</BASE_COORD>
</READ_SPEC>
<READ_SPEC>
<READ_INDEX>1</READ_INDEX>
<READ_CLASS>Application Read</READ_CLASS>
<READ_TYPE>Reverse</READ_TYPE>
<BASE_COORD>99</BASE_COORD>
</READ_SPEC>
</SPOT_DECODE_SPEC>
</SPOT_DESCRIPTOR>
So this run is supposed to have (according to the metadata) 196bp of data, split into 2x98bp reads. We can start to see where the issue arises therefore. The fastq-dump file knows that read2 is supposed to start at position 99, so it splits its read string there, but because there are only 99bp instead of 198 we get a very truncated read 2.
There are two possible reasons for this problem therefore, either there is read data missing, or the metadata is wrong. Since this is an RNA-Seq run we can use the properties of the data to try to distinguish these two options.
A simple check is whether the same data comes down from a different repository. We can pull the fastq files from ENA. If we do this we again find a 98bp read and a 1bp read, so at least the problem is consistent.
One of the notable things about RNA-Seq data is that due to biased priming you get sequence composition bias at the start of each read. If the data we have contains both read 1 and read 2 we should therefore see this bias at the start of the data, but see it again in the middle. When we run fastqc on the unsplit data we see the following:
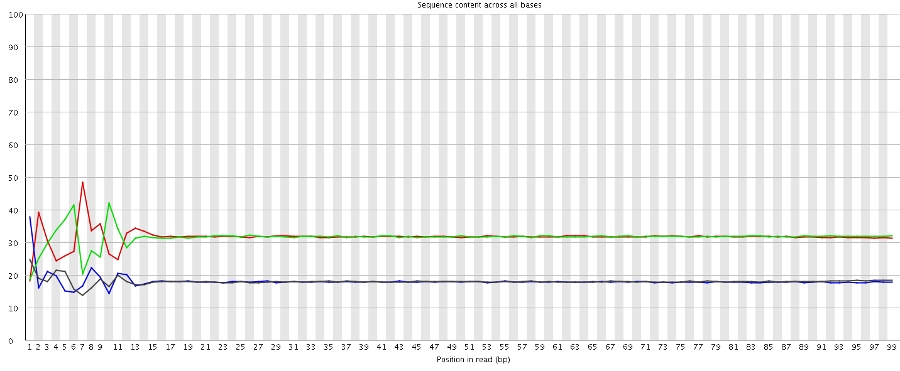
So there is no evidence for a second set of biased positions, suggesting that we’re simply missing data. Interestingly we don’t even see a bias in the last position which suggests that the 99th base may not be the first base of the second read either.
Going back to the original paper we find the material and methods say:
10986225 reads; of these: 10986225 (100.00%) were unpaired; of these: 1142174 (10.40%) aligned 0 times 8766188 (79.79%) aligned exactly 1 time 1077863 (9.81%) aligned >1 times 89.60% overall alignment rate
Mitigation
In this case we can proceed using just the read1 data. We can pursue the rest of the data with the sequence repositories and the original authors to try to get a more complete dataset.
Prevention
This type of error should really be prevented during the submission process to the public repositories. Some simple data validation would have shown that the available data could not possibly match the metadata and yet two repositories provide this invalid data for download.
Lessons Learnt
I guess the message here is don’t automatically trust data from public repositories, and always try to validate that data and metadata match. You should also not always rely on program which fail (such as fastq-dump here) actually throwing an error. The exit code for the extraction was 0 in all cases so it wasn’t caught by our normal monitoring.
Datasets
The accession used here is SRR2133898 and the data shown was from 3 Mar 2016. It’s possible that this entry will be corrected in future if the full dataset is submitted.
4 thoughts on "Data can be corrupted upon extraction from SRA files"
Antonio Rodriguez Franco
Another issue I had is the extraction of not synchronized paired end data. Some SRA data has missing mate reads possibly due to a stringent or inadequate quaility trimming
In this case, it is advisable to use fastq-dump with the –split-3 legacy command, that renders two fully synchronized paired files (expected by many other programs) and a third of orphans reads
Simon Andrews
Thanks for sharing that – that’s not one we’ve ever seen. Do you have an example accession you can share?
I’m kind of surprised that this would happen in the database as the internal storage for SRA files concatenates the reads together and then only splits them on extraction. In this case it might not be the databases fault, but the submitter messing up the processing of their samples before submitting them (even though they shouldn’t be submitting trimmed or filtered data anyway).
Norman Warthmann
i’ve seen this: e.g. SRR1261867
Chris Jacobs
Thanks for this post. Also the tip by Antonio was very helpful. I came across SRA data that has 0-length reads inserted in the files to ‘complete’ pairs. However, fastq-dump removes them and that screws up the order which prevents paired-end aligning with for example hisat2, which depends on the order of the pairs being the same in the left and right file. With the –split-3 option it ‘saves’ the pairing by outputting the orphaned reads to a 3th file.
Example (sra toolkit v2.8.2):
> fastq-dump -A SRR1554537 –gzip –split-3
Rejected 11724434 READS because READLEN < 1
Read 66858380 spots for SRR1554537
Written 66858380 spots for SRR1554537"
Comments are closed