Introduction
In the analysis of RNA-Seq data we often make an implicit assumption about our knowledge of the transcriptome in the species we’re working in. In some species the content of the transcriptome is very well understood and that information can be used to aid mapping. Some mapping programs such as Kallisto only map to a transcriptome and ignore reads which would map elsewhere in the genome. Other mappers such as TopHat employ a dual strategy where data is initially mapped to a transcriptome, and only goes on to map against the entire genome should no match be found within the transcriptome.
There is however a problem with these types of approach which revolves around the ability to define a read as uniquely mapping. For many analysis strategies it is easier and more accurate to deal only with reads which uniquely map to the reference so you can be sure that you are correctly assigning your signal. When mapping to a transcriptome though it is possible to have a read map uniquely within the transcriptome, but have it able to map to very many locations within the genome had such a mapping been performed. In many libraries these are purely theoretical concerns as the number of these reads is generally low, but where a significant amount of non-geneic transcription is present, or where DNA contamination occurs then these reads can drastically affect an otherwise robust analysis.
The Symptoms
This sort of artefact generally does not produce obvious symptoms initially. Overall metrics for the QC of the sample will not generally be adversely affected since the bias affects only a small number of genes. Even reviewing the data manually may not identify the problem. The problem is only really evident when reviewing some of the signficantly differentially expressed genes and looking at the distribution of the reads over the transcript. Normally you would expect that the reads would be relatively evenly distributed over the whole area of the transcript as shown below:
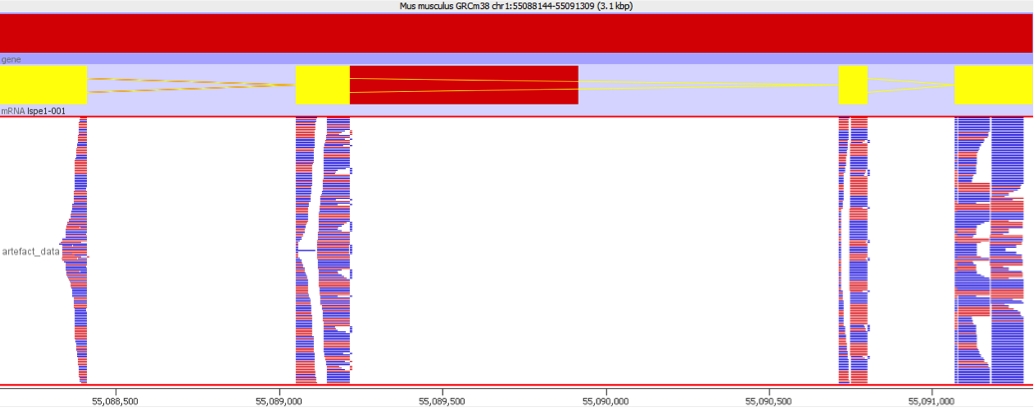
In a gene affected by this artefact the coverage would look very different, with a large number of reads mapping to a very limited region of the gene body. This would be the region which contained the repetitive sequence which is the underlying cause of this issue.
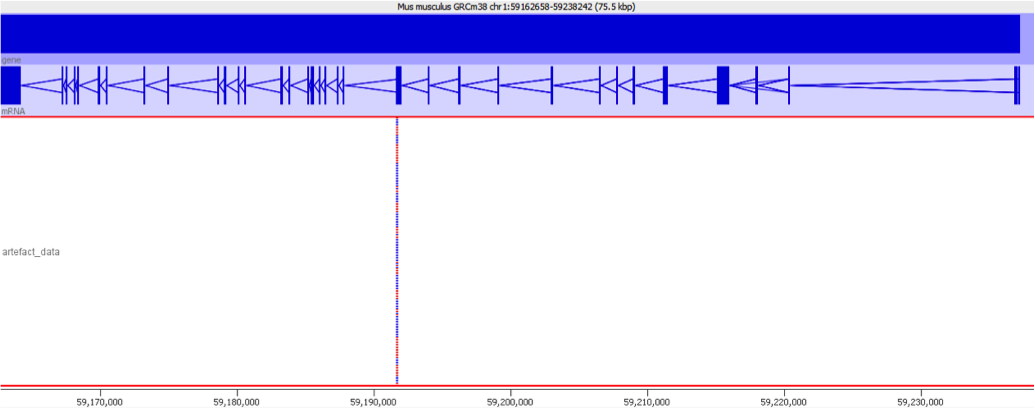
Diagnosis
When we encountered this issue we worked out the underlying cause by putting some of the aberrantly mapping reads through mapping with different mappers. We found that using a mapper which did not do an initial mapping to the transcriptome completely removed the signal which caused these genes to be selected initially.
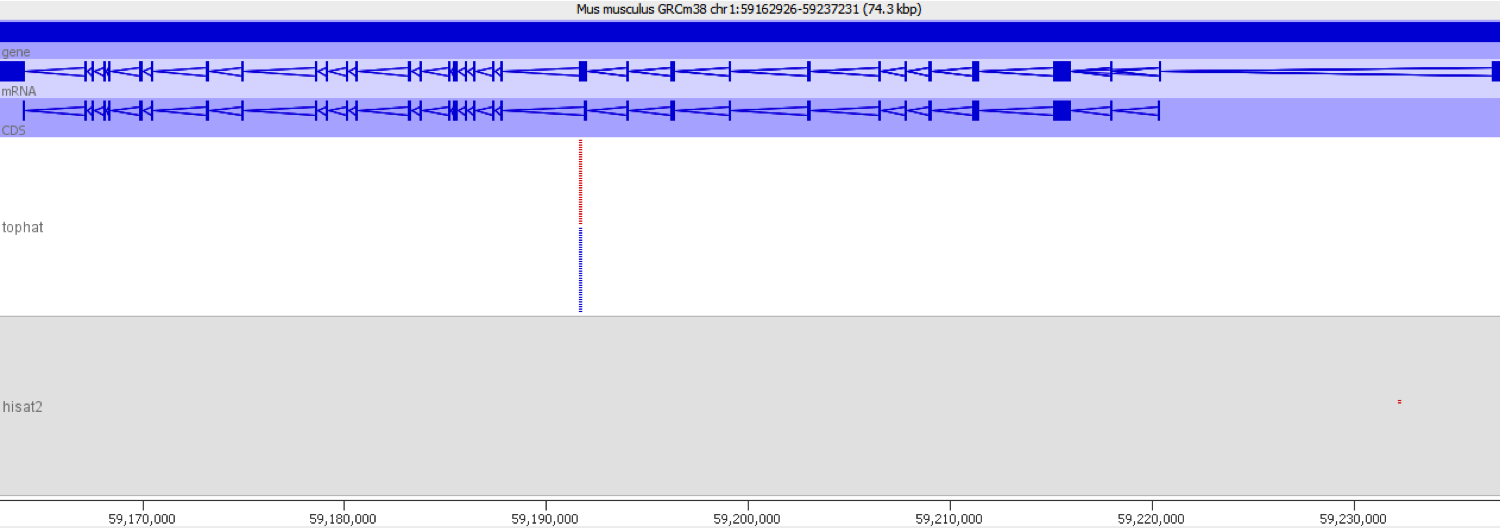
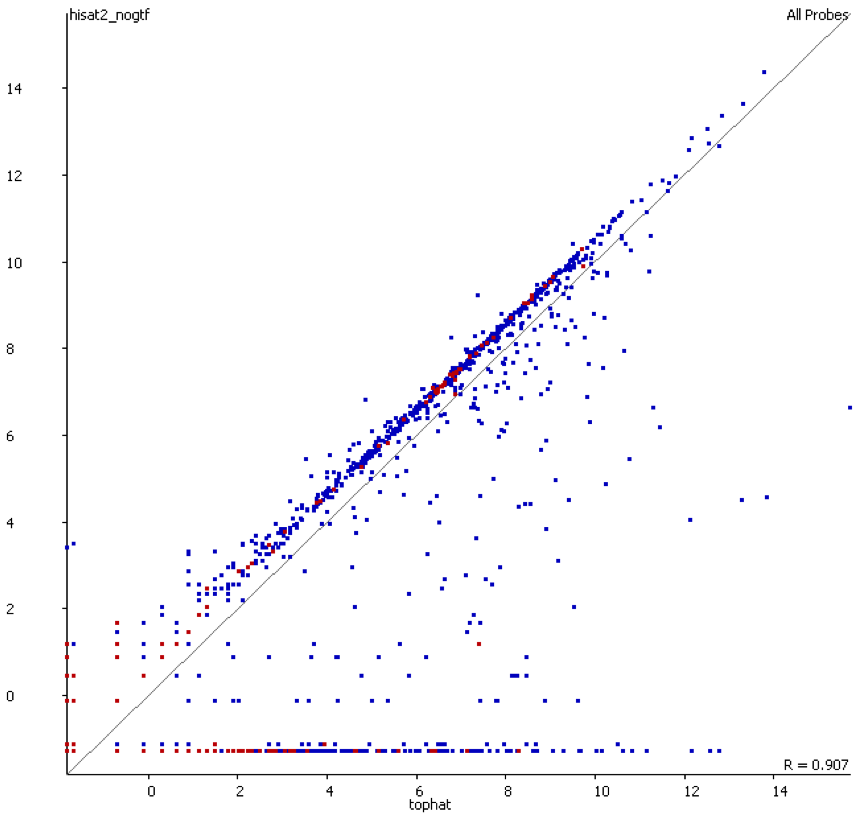
We also compared the quantitation produced by the two mappers used and found that there was a consistent bias for the transcriptome-first mapper to overestimate the expression relative to the whole genome mapper.
Mitigation
This issue was mitigated by re-mapping the data with the genome-first mapper and using this new mapper for the remainder of our RNA-Seq data.
Lessons Learnt
It’s always worth reviewing the hits you get from an analysis pipeline back through as far as the raw data to ensure that the signal you see comes in a form you would expect.
Software
The plots show here were all generated in SeqMonk.
The mapper which generated the artefacts was TopHat, the issue was reported to the TopHat developers, but the nature of the problem is such that there isn’t really a way to fix it without severely impacting the performance of the program.
The mapper which removed the artefacts was Hisat2, but we would expect that any other whole genome RNA-Seq mapper would have produced similar results.
One thought on "Mapping to a transcriptome can incorrectly report reads as mapping uniquely"
Martin Smith
Another way to avoid this artifact is to perform ab initio transcriptome assembly on the sequencing data (provided it is deep enough). The resulting transcriptome is then used as a sample-specific reference, which routinely yields read mapping rates over 85-90%. This circumvents the issue of forcing reads to align to a generic reference transcriptome that contains genes not present in the surveyed sample (or sample-specific genes that are not annotated, like lincRNAs). For example, an RNAseq sample from a kidney will be much less complex than one from brain. Less ambiguity in the quantification stage means more accurate differential expression.
In the example above, the contaminating repetitive DNA is filtered out when mapping the assembled transcriptome to the genome (with BLAT for instance). Most times, full-length or partially assembled transcripts map uniquely to a reference genome, with the exception of fusion genes and (to a lesser extent) pseudogenes. Full-length transcripts that map to more than a few regions can be ignored.
Comments are closed