Introduction
Recent years have seen ever improving DNA sequencers, offering greater depths of coverage and faster processing times. A recent innovation by Illumina was the development of ordered flow cells, debuting on the HiSeq X and subsequently carried over onto the HiSeq 3000 and 4000 systems. This patterned arrangement comprised billions of fixed-sized nanowells distributed uniformly across a sequencing flow cell, enabling the discrimination of separate DNA clusters at much higher densities than possible on previous generation machines. Despite these advances, there is evidence that patterned flow cells generate more duplicate reads than the earlier designs.
The Symptoms
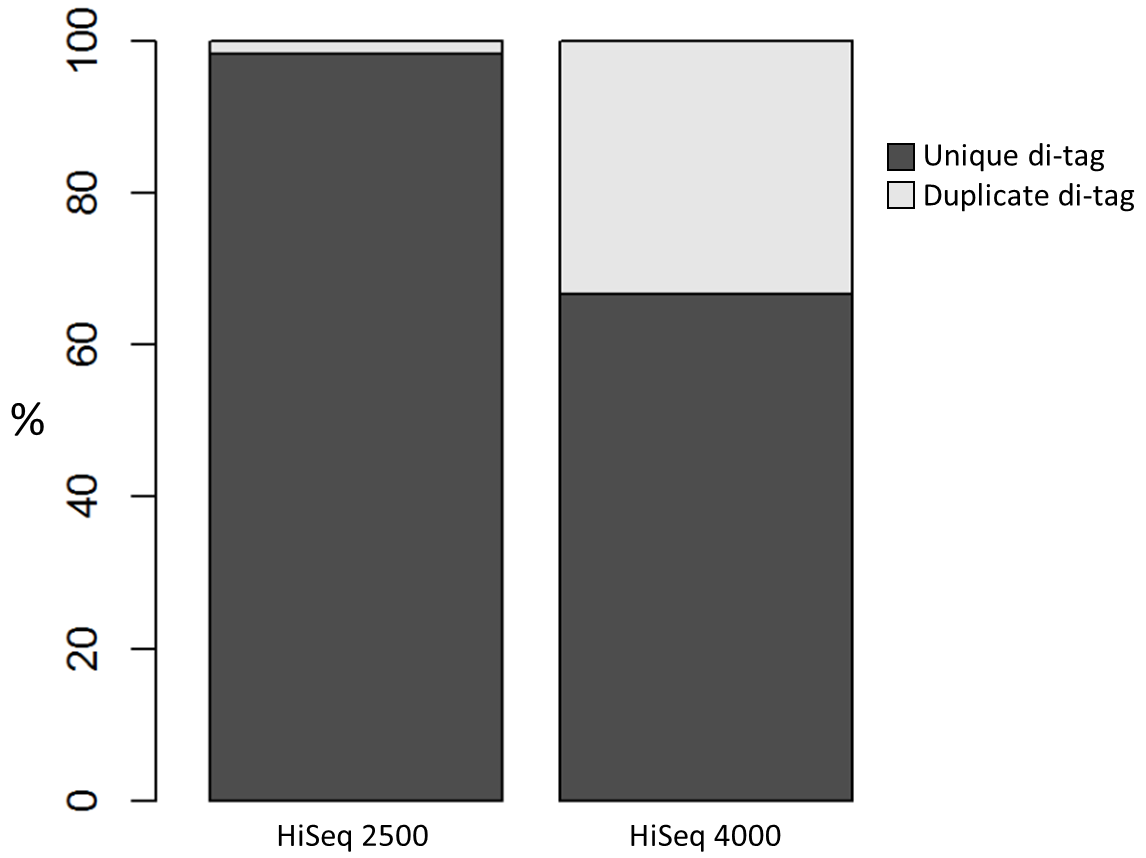
We recently used a HiSeq 4000 and a HiSeq 2500 – which uses a conventional flow cell – to sequence the same Hi-C library. (Hi-C is a technique used to determine the three-dimensional conformation of a genome. This is achieved by performing paired-end sequencing of Hi-C di-tags, which comprise two DNA fragments ligated together. These fragments derive from different genomic positions that were in spatial proximity in the nucleus (Lieberman-Aiden et al.).) The sequencing data were processed using HiCUP, a pipeline tailored for mapping and removing artefacts from Hi-C FASTQ files (Wingett et al.). As expected, the HiSeq 4000 generated far more raw reads (383M) than the HiSeq 2500 (257M). Surprisingly however, the proportion of duplicates differed substantially between the two sequencing runs of the same library. Only 2% of the HiSeq 2500 di-tags were removed after de-duplicating, in stark contrast to the 33% removed from the data generated by the HiSeq 4000. The effect of duplicate removal was to leave both samples with 55M reads at the end of the HiCUP pipeline, thereby offsetting the initial gain made by the increased read output of the HiSeq 4000.
Diagnosis
Before investigating the differences between the two machines, we wanted to rule out the possibility that the increased proportion of duplicates detected by the HiSeq 4000 was simply a result of sequencing more di-tags – i.e. the more a sample was sequenced the higher the probability that any given read was a duplicate. To check for this, the HiSeq 4000 FASTQ file was randomly downsized to the same number of reads as found in the file generated by the HiSeq 2500. During HiCUP processing 25% of the di-tags were now discarded during the de-duplication step, still far more than the 2% discarded than when processing the HiSeq 2500 data.
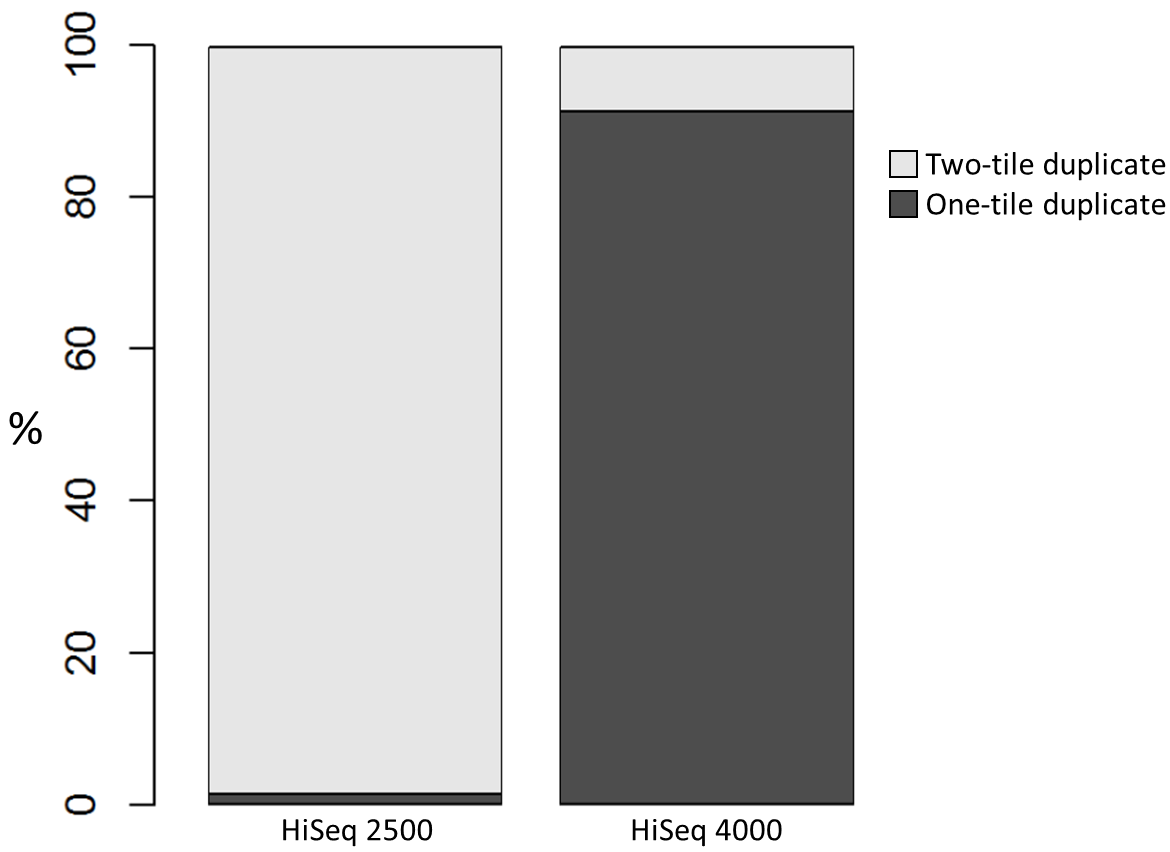
To investigate the possible cause of the duplication we analysed the spatial distribution of duplicate di-tags on the flow cells. For both machines the duplicates were scattered in a uniform manner and did not show significant duplication “hotspots”. While duplicates were not localised to particular regions of a flow cell, it was still possible that, in general, duplicate di-tags may have co-localised with their exact copies. To test this hypothesis we identified di-tags present in two copies and recorded whether they mapped to one or two tiles (each Illumina flow cell comprises multiple tiles). Significantly, 1% of HiSeq 2500 duplicates comprised di-tags originating from the same tile. In contrast, 92% of the duplicate pairs were located on a single tile for the HiSeq 4000. This close proximity suggest that the duplicates observed on the HiSeq 4000 were largely machine-specific artefacts.
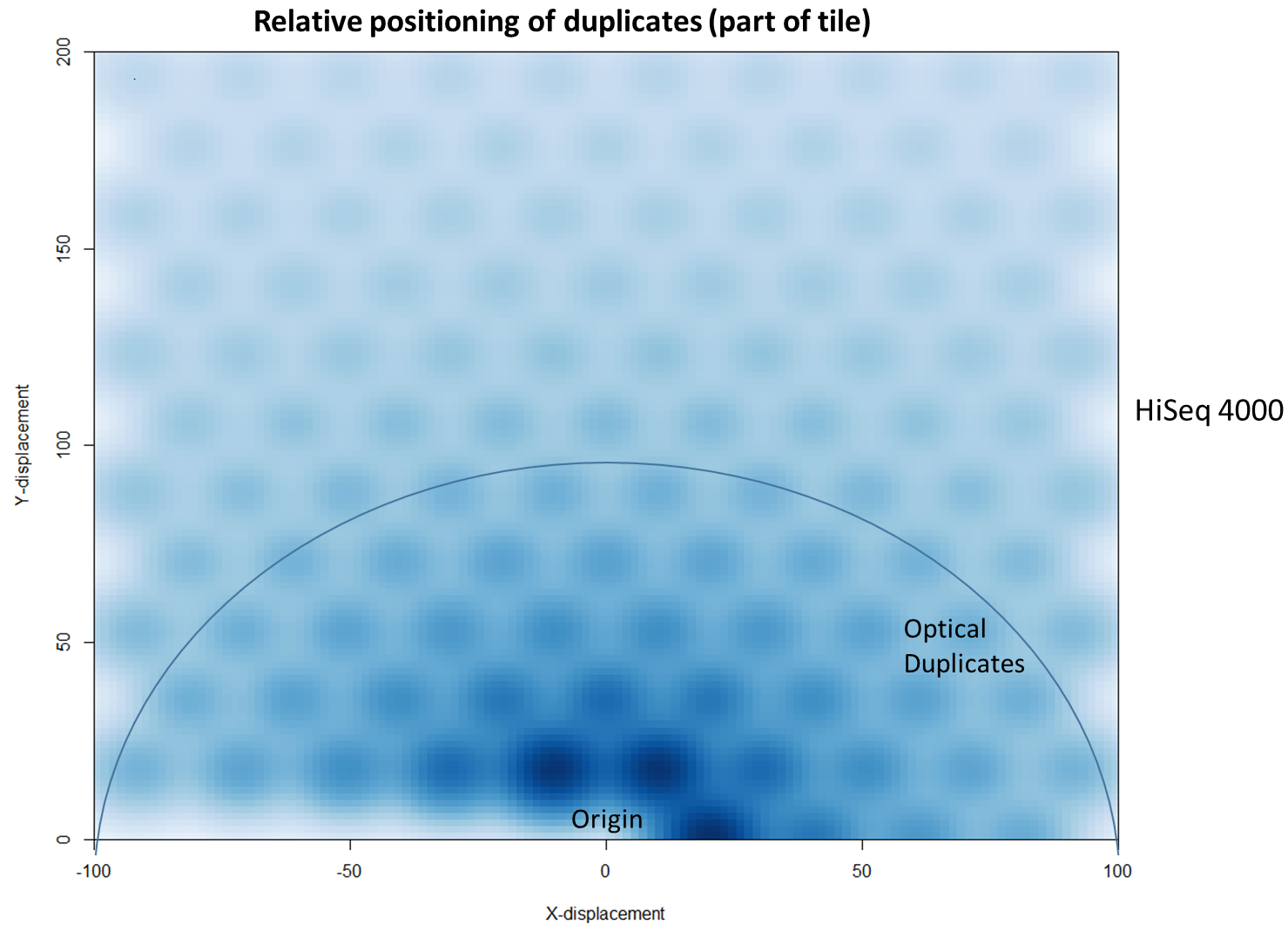
To further characterise this two-dimensional separation, we extracted duplicates which localised to only one tile and then recorded the relative position of a di-tag to its exact duplicate (this is possible because FASTQ files record the coordinates of each cluster). The figures below show these findings as density plots (for each di-tag, one read was specified as the origin, and the chart shows the relative position of the “other ends” to the origin).
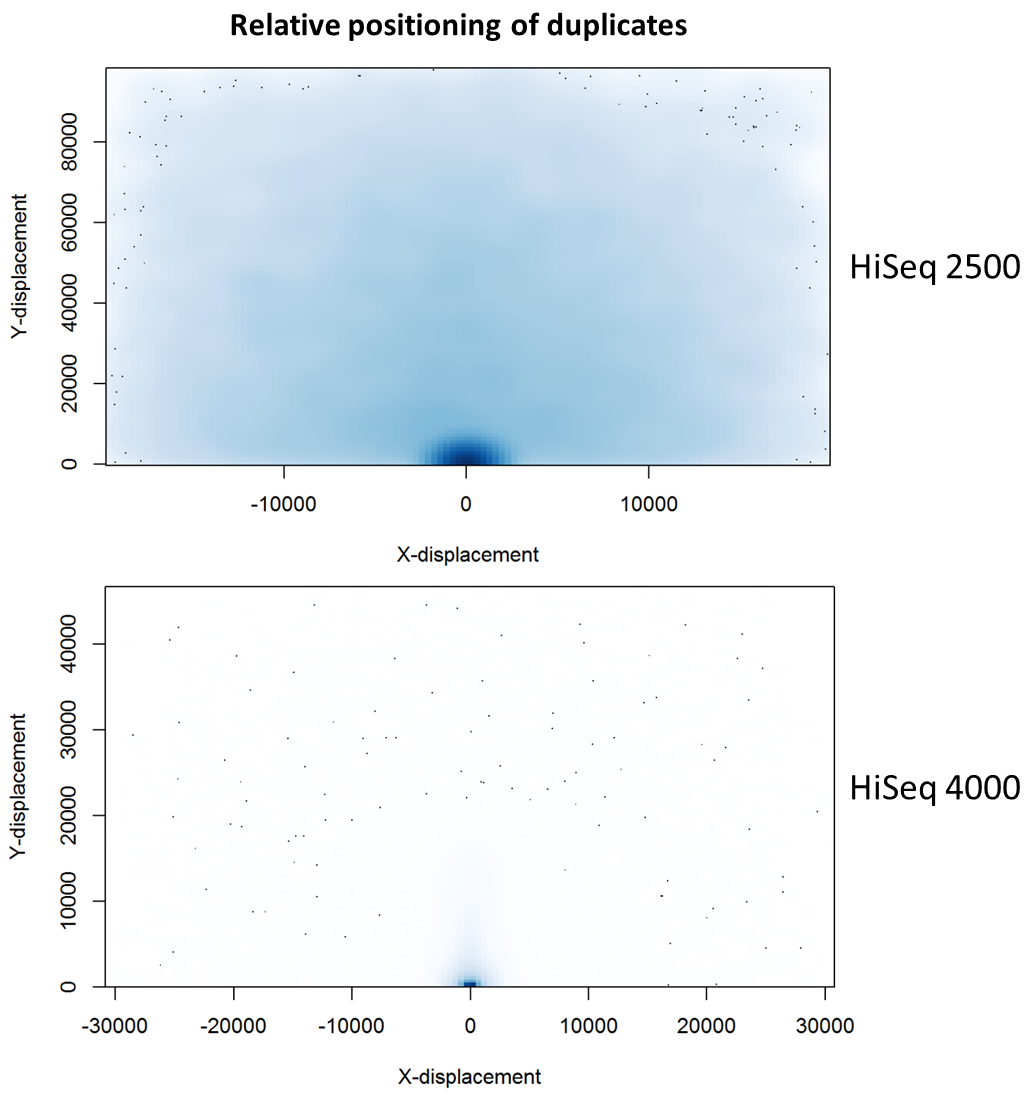
For the HiSeq 2500 there is, in general, a uniform distribution across the plot, except for a high-density region in close proximity to the origin. This elevated density around the origin is much more pronounced when analysing the HiSeq 4000 data, in which almost all the other-ends localise to this region. We hypothesise that the other-ends positioned far alway from the origin are true biological duplicates or experimental PCR duplicates. In contrast, those other-ends close to the origin are more likely to be generated by the machine itself. Again, this is indicative of the HiSeq 4000 generating more duplication artefacts.
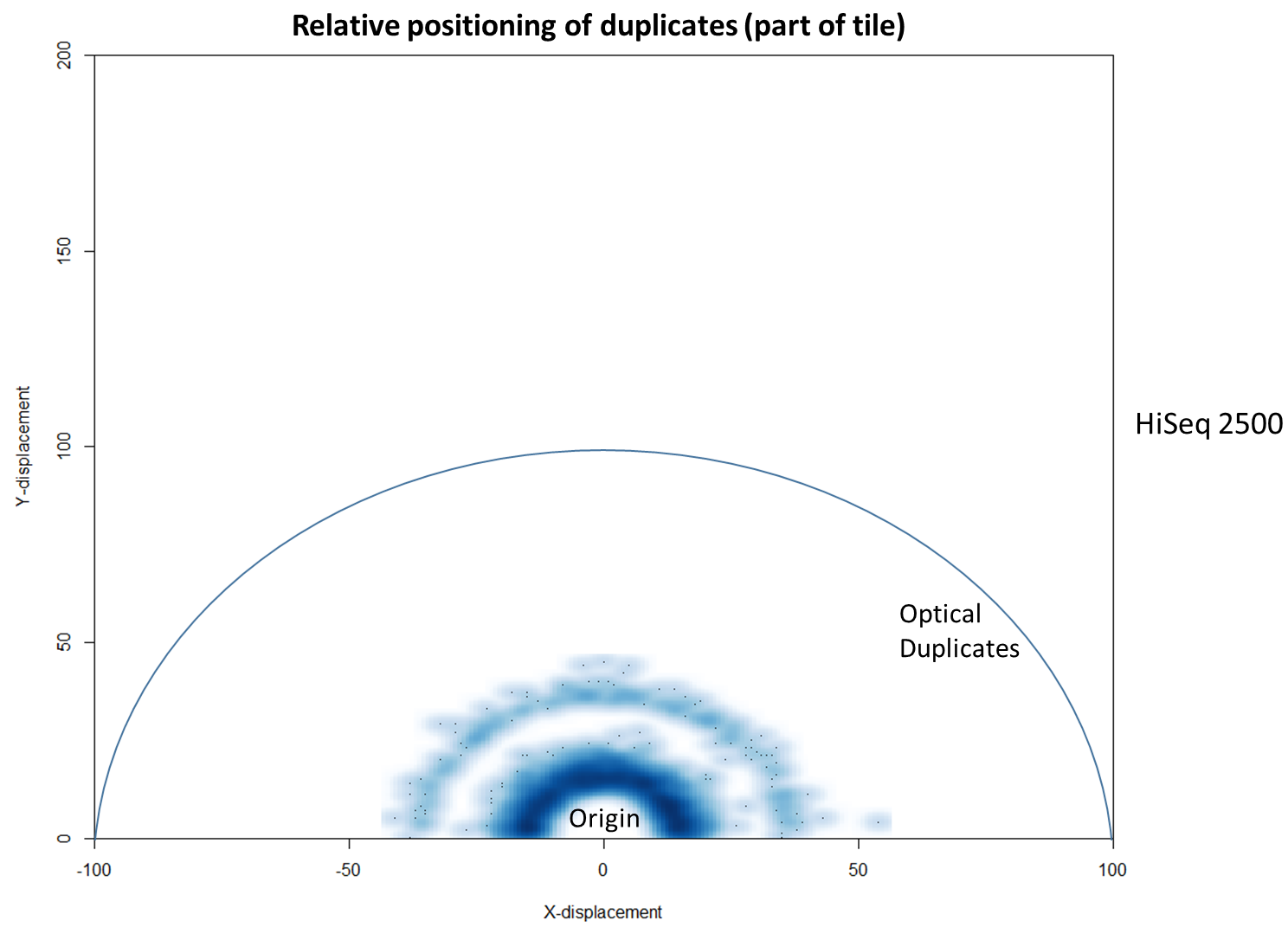
We then investigated whether such duplicates on the HiSeq 4000 were confined to adjacent nanowells, or multiple nanowells in the same local region of a flow cell. Although we were unable to obtain direct information relating the FASTQ coordinate system to individual nanowells, it was possible, by creating a density plot of the region immediately around the origin, to visualise the ordered array of the HiSeq 4000 flow cell. The plot clearly shows that duplicates are found in multiple wells around the origin, and this trend decreases as one moves from the origin. Also shown below is the same plot, but of the HiSeq 2500 data. As expected no nanowell pattern is visible.
Mitigation
Considering the vast potential combination of Hi-C interactions, it should be expected a priori that exact sequence duplicates are more likely artefacts than independent Hi-C events, which has indeed been shown to be the case (Wingett et al.). (Indeed, sequencing Hi-C libraries presents a good method to quantify levels of experimental duplication.) Consequently, duplicates may be identified and removed as part of a standard Hi-C bioinformatics pipeline. This is important since duplicates which do not reflect an underlying biological trend may not only reduce the potential complexity of a data set, they may lead to incorrect inferences being drawn in the post-sequencing analysis. This is a particular problem if certain DNA molecules are inherently more likely than others to form duplicates. For example, it was suggested to us that DNA fragment length may affect duplication rate (in fact we found no evidence for this on further inspection of the data, but nevertheless the potential problem of duplication bias still stands).
Although it is relatively straight-forward to remove duplicates from a mapped dataset, this is not always desirable when genome coverage is high and exact duplicates should be expected, as often observed in RNA-Seq experiments. With the knowledge that patterned-flow cells produce duplicates which are positioned close together, it should be possible to model the separation of such artefacts and set a “proximity filter” to selectively remove them. The histogram below shows the frequencies of the separation distances between duplicates generated on the HiSeq 4000.
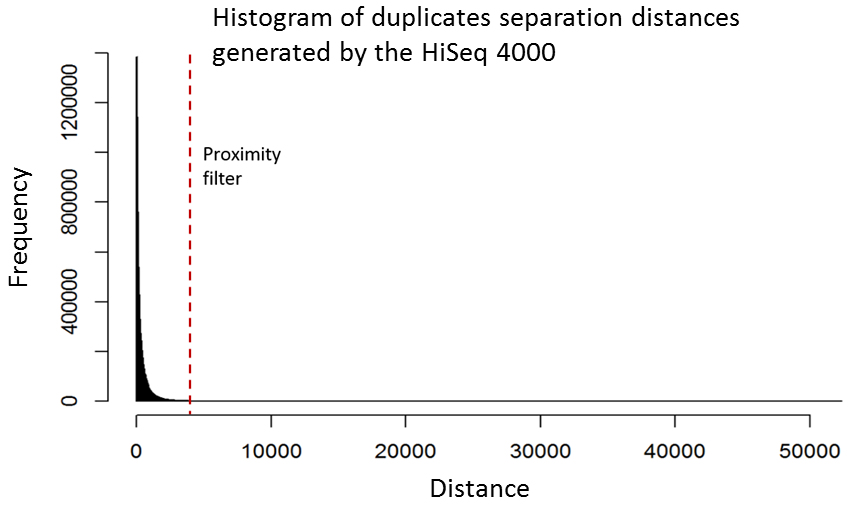
In fact, there are tools already available to identify and label duplicates positioned extremely close together, and these are termed “optical duplicates”. Such duplicates are already known to occur on unpatterned flow cells and result from optical sensor artefacts. Picard is commonly used bioinformatics software than can identify such duplicates. However, by default the program expects duplicates to be within 100 pixels (as illustrated on the density plots above). Although this filter does remove the intense signal around the origin for unordered flowcells, it does not however eliminate ordered flow cell duplicates. In light of this, the Picard documentation recommends increasing the filter to remove duplicates separated by a distance less than 2500 pixels when analysing data generated on a patterned flow cell. This is broadly in agreement with the histogram above and should remove most machine-generated artefacts
While this proximity filtering strategy does mitigate the problem when applied as an “in-house” sequencing/bioinformatics pipeline, it will not be applicable to data submitted to public repositories in which FASTQ header information (which records the cluster co-ordinates) has been removed. Consequently, conclusions reached when processing such third-party data may incorrect owing to duplication biases.
Prevention
The increased propensity for duplicate generation on patterned flow cells has been discussed previously by other researchers. In a recent article published on the CRUK-CI Core Genomics blog, James Hadfield reported that library molecules from a cluster may return to the surrounding solution and then act as a seed for second cluster in a nearby flow cell. It was proposed that these re-seeding events may be minimised by increasing the loading concentration of a library, although this would be at the expense of creating unusable polyclonal clusters. Consequently, a balance would need to be made between these two competing problems to maximise the number of unique valid DNA sequence reads. We discussed this directly with Illumina who agreed with this strategy and suggested that patterned flowcell machines may need to be calibrated on a sequencer-by-sequencer basis to determine optimal loading concentrations.
Lessons Learnt
While patterned flow cells, as seen on the HiSeq 4000, generate a substantially increased number of reads, there is increased scope for duplicate generation. It appears the patterned design of the flow cell, intended to increase discrimination between tightly packed clusters, may itself lead to the generation of duplicates. Such duplicates lead to reduced sequencing depth and the potential to skew experimental results. To ameliorate this situation requires further experimental preparation and possibly sequencer-specific bioinformatics filtering of results.
References
Lieberman-Aiden, Erez et al. “Comprehensive Mapping of Long Range Interactions Reveals Folding Principles of the Human Genome.” Science (New York, N.Y.) 326.5950 (2009): 289–293. PMC. Web. 13 Feb. 2017.
Wingett, Steven et al. “HiCUP: Pipeline for Mapping and Processing Hi-C Data.” F1000Research 4 (2015): 1310. PMC. Web. 13 Feb. 2017.
Software
HiCUP, using defaults but retaining intermediate BAM files that contained putative duplicate di-tags. HiCUP used Bowtie 2 as the sequence aligner.
Picard: Java tools for manipulating sequencing data and formats such as SAM/BAM/CRAM and VCF.
10 thoughts on "Illumina Patterned Flow Cells Generate Duplicated Sequences"
Yannick
Thank you very much for the detailed analysis and plots- this is extremely informative.
Tom Smith
Very nice analysis. I really appreciate the detailed walk-through of the steps taken to diagnose the issue. Do you know if there is a generic tool to make the histogram for duplicates vs. distance you include above? If not, perhaps you could suggest this to the Picard team? I think this would be very useful if optical duplicates are going to become more of an issue with illumina sequencing. Did illumina say they were working to improve the pattern flow cells to reduce the problem?
With regards to what to do about possible PCR duplicates when you expect to observe reads with the same coordinates, e.g RNA-Seq, I’d recommend including Unique Molecular Identifiers (UMIs). This can be done at very little additional cost and enables removal of PCR duplicates whilst retaining truly distinct molecules. We’ve recently published UMI-tools for error-aware removal of PCR duplicates using UMIs (http://genome.cshlp.org/content/early/2017/01/18/gr.209601.116)
Steven Wingett
I am not aware of any generic tool, although we are considering adding duplicate characterisation to our FastQC software. I do not know what steps Illumina are taking to deal with the problem.
I agree that UMIs should help identify such duplicates.
Geno Max
We have discussed this extensively in a thread over at SeqAnswers: http://seqanswers.com/forums/showthread.php?t=72901&highlight=clumpify&page=3
Brian Bushnell has a new tool called “clumpify” that is part of the BBTools package that can mark/remove these duplicates without requiring alignments.
Brian Bushnell
I’d like to note the Clumpify can do duplicate-detection, marking, and removal, as shown here: https://www.biostars.org/p/229842/
It’s helpful in that it can do this at the beginning of a pipeline without mapping, which is necessary in situations when you are doing denovo assembly and thus don’t have a reference; additionally, it makes pipelines more efficient to remove duplicates at the very beginning. I’d suggest that overall, reference-free duplicate detection is probably less biased than mapping-based duplicate detection, not to mention that it’s far faster.
Jorge González-Domínguez
Thank you for the analysis! Since its version 2.0.5 the ParDRe tool (https://academic.oup.com/bioinformatics/article-lookup/doi/10.1093/bioinformatics/btw038) provides an option to only remove optical duplicates with separation distance lower than a threshold (parameter -d) and leave the other duplicates or near-duplicates.
It is publicly available here: https://sourceforge.net/projects/pardre
Judith
Hi, very nice article. Would it be possible to obtain a doi, may be by putting it on biorxiv? I’d like to be able to cite it properly.
Steven Wingett
Thanks for the kind words. We view these posts more as news stories rather than research articles as so at present we do not intend to deposit on biorxiv. Hopefully the URL will be sufficient for your purposes.
Andrea Bours
Hello,
Thank you so much for this investigation into duplicates and the platforms that were used to produce reads.
I’m hoping that you could enlighten me and probably others who are having the same question.
Illumina has launched new sequencing technology the novaseq and the patterned flowcells are even more densely spaced to each other. Do you have any recommendation on Picard Markduplicates on pixel distance setting?
Some forums suggest 12000 instead of the 2500 for hiseq, do you agree with this?
Thank you and best,
Steven Wingett
Thanks for your message. It certainly would be interesting to know whether and to what extent this artefact affects NovaSeq runs. We do not have such a machine at my institute, and consequently I have never looked at Hi-C data run on this sequencer. I imagine the issue still exists, but I am unable to give specifics as to the duplication proximity threshold.
Comments are closed